 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7oa9 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Human Menin in complex with Fragment 21 | ||||||
 Components Components | Isoform 2 of Menin | ||||||
 Keywords Keywords | ONCOPROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of cyclin-dependent protein serine/threonine kinase activity / Y-form DNA binding / MLL1/2 complex / osteoblast development / negative regulation of JNK cascade / T-helper 2 cell differentiation / Formation of WDR5-containing histone-modifying complexes / positive regulation of transforming growth factor beta receptor signaling pathway / negative regulation of protein phosphorylation / histone methyltransferase complex ...negative regulation of cyclin-dependent protein serine/threonine kinase activity / Y-form DNA binding / MLL1/2 complex / osteoblast development / negative regulation of JNK cascade / T-helper 2 cell differentiation / Formation of WDR5-containing histone-modifying complexes / positive regulation of transforming growth factor beta receptor signaling pathway / negative regulation of protein phosphorylation / histone methyltransferase complex / R-SMAD binding / negative regulation of cell cycle / cleavage furrow / MLL1 complex / negative regulation of osteoblast differentiation / RHO GTPases activate IQGAPs / negative regulation of DNA-binding transcription factor activity / response to UV / four-way junction DNA binding / transcription repressor complex / transcription initiation-coupled chromatin remodeling / Deactivation of the beta-catenin transactivating complex / SMAD2/SMAD3:SMAD4 heterotrimer regulates transcription / response to gamma radiation / Post-translational protein phosphorylation / phosphoprotein binding / Formation of the beta-catenin:TCF transactivating complex / nuclear matrix / Regulation of Insulin-like Growth Factor (IGF) transport and uptake by Insulin-like Growth Factor Binding Proteins (IGFBPs) / MAPK cascade / double-stranded DNA binding / protein-macromolecule adaptor activity / chromosome, telomeric region / transcription cis-regulatory region binding / endoplasmic reticulum lumen / negative regulation of cell population proliferation / DNA repair / negative regulation of DNA-templated transcription / DNA damage response / chromatin binding / regulation of transcription by RNA polymerase II / chromatin / negative regulation of transcription by RNA polymerase II / positive regulation of transcription by RNA polymerase II / protein-containing complex / nucleoplasm / nucleus / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.098 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.098 Å | ||||||
 Authors Authors | Groves, M.R. / Gao, K. | ||||||
 Citation Citation | Journal: To Be Published Title: Crystal structure of Human Menin in complex with Fragment 21 Authors: Groves, M.R. / Gao, K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7oa9.cif.gz | 111.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7oa9.ent.gz | 81.5 KB | Display | PDB format |
| PDBx/mmJSON format | 7oa9.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 7oa9_validation.pdf.gz | 718.5 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 7oa9_full_validation.pdf.gz | 723.1 KB | Display | |
| Data in XML | 7oa9_validation.xml.gz | 19.8 KB | Display | |
| Data in CIF | 7oa9_validation.cif.gz | 28.7 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/oa/7oa9ftp://data.pdbj.org/pub/pdb/validation_reports/oa/7oa9 | HTTPS FTP |
-Related structure data
| Related structure data |  5ddcS S: Starting model for refinement |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 56033.516 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: MEN1, SCG2 / Production host:  | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-V6K / (~{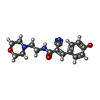  Mass: 301.340 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H19N3O3 / Feature type: SUBJECT OF INVESTIGATION Mass: 301.340 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H19N3O3 / Feature type: SUBJECT OF INVESTIGATION | ||||||
| #3: Chemical | ChemComp-EDO /   Mass: 62.068 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C2H6O2#4: Chemical | ChemComp-GOL / |   Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 169 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 169 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.84 Å3/Da / Density % sol: 56.65 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 7.5 / Details: 12% PEG 3350 14% Glycerol / PH range: 7.5-8.0 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: PETRA III, DESY  / Beamline: P11 / Wavelength: 1.0332 Å / Beamline: P11 / Wavelength: 1.0332 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Nov 30, 2019 |
| Radiation | Monochromator: Si 111 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.0332 Å / Relative weight: 1 |
| Reflection | Resolution: 2.098→49 Å / Num. obs: 37566 / % possible obs: 99.93 % / Redundancy: 17.6 % / Biso Wilson estimate: 36.81 Å2 / CC1/2: 0.999 / CC star: 1 / Rmerge(I) obs: 0.09 / Rrim(I) all: 0.09 / Net I/σ(I): 22 |
| Reflection shell | Resolution: 2.098→2.173 Å / Redundancy: 12 % / Rmerge(I) obs: 0.582 / Num. unique obs: 3674 / CC1/2: 0.957 / CC star: 0.989 / Rrim(I) all: 0.599 / % possible all: 99.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5DDC Resolution: 2.098→48.21 Å / SU ML: 0.21 / Cross valid method: THROUGHOUT / σ(F): 1.34 / Phase error: 23.86 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 103.27 Å2 / Biso mean: 46.261 Å2 / Biso min: 21.24 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.098→48.21 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0
|
