 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7nwe: CRYSTAL STRUCTURE OF HUMAN CYTOSOLIC BRANCHED-CHAIN AMINOTRANSFER... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7nwe | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF HUMAN CYTOSOLIC BRANCHED-CHAIN AMINOTRANSFERASE (BCAT1) IN COMPLEX WITH PLP AND INHIBITOR COMPOUND 10 | ||||||
 Components Components | Branched-chain-amino-acid aminotransferase, cytosolic | ||||||
 Keywords Keywords | TRANSFERASE / BRANCHED-CHAIN-AMINO-ACID AMINOTRANSFERASE / PYRIDOXAL-PHOSPHATE-DEPENDENT AMINOTRANSFERASE / SMALL MOLECULE INHIBITOR | ||||||
| Function / homology |  Function and homology information Function and homology informationbranched-chain-amino-acid:2-oxoglutarate transaminase activity / L-valine:2-oxoglutarate transaminase activity / L-isoleucine:2-oxoglutarate transaminase activity / L-leucine:2-oxoglutarate transaminase activity / branched-chain amino acid biosynthetic process / branched-chain-amino-acid transaminase / L-leucine biosynthetic process / Branched-chain amino acid catabolism / branched-chain amino acid catabolic process / L-valine biosynthetic process ...branched-chain-amino-acid:2-oxoglutarate transaminase activity / L-valine:2-oxoglutarate transaminase activity / L-isoleucine:2-oxoglutarate transaminase activity / L-leucine:2-oxoglutarate transaminase activity / branched-chain amino acid biosynthetic process / branched-chain-amino-acid transaminase / L-leucine biosynthetic process / Branched-chain amino acid catabolism / branched-chain amino acid catabolic process / L-valine biosynthetic process / lipid metabolic process / G1/S transition of mitotic cell cycle / mitochondrion / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.54 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.54 Å | ||||||
 Authors Authors | Hillig, R.C. | ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2022 Title: BAY-069, a Novel (Trifluoromethyl)pyrimidinedione-Based BCAT1/2 Inhibitor and Chemical Probe. Authors: Gunther, J. / Hillig, R.C. / Zimmermann, K. / Kaulfuss, S. / Lemos, C. / Nguyen, D. / Rehwinkel, H. / Habgood, M. / Lechner, C. / Neuhaus, R. / Ganzer, U. / Drewes, M. / Chai, J. / Bouche, L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7nwe.cif.gz | 301.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7nwe.ent.gz | 244.9 KB | Display | PDB format |
| PDBx/mmJSON format | 7nwe.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/nw/7nweftp://data.pdbj.org/pub/pdb/validation_reports/nw/7nwe | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7ntrC  7nwaC  7nwbC  7nwcC  7nwmC  7nxnC  7nxoC  7ny2C  7ny9C  7nyaC  2abjS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||
| Unit cell |
| ||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: _ / Ens-ID: 1 / Beg auth comp-ID: GLY / Beg label comp-ID: GLY / End auth comp-ID: LEU / End label comp-ID: LEU / Refine code: _ / Auth seq-ID: 23 - 385 / Label seq-ID: 26 - 388
|
-Components
| #1: Protein | Mass: 43290.312 Da / Num. of mol.: 2 / Mutation: Ser379Arg Source method: isolated from a genetically manipulated source Details: SYNONYM: BCAT(C), BCAT1 / Source: (gene. exp.) Homo sapiens (human) / Gene: BCAT1, BCT1, ECA39 / Production host:  References: UniProt: P54687, branched-chain-amino-acid transaminase #2: Chemical | 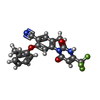  Mass: 401.339 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H14F3N3O3 / Feature type: SUBJECT OF INVESTIGATION Mass: 401.339 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H14F3N3O3 / Feature type: SUBJECT OF INVESTIGATION#3: Chemical |   Mass: 247.142 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H10NO6P Mass: 247.142 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H10NO6P#4: Chemical | ChemComp-MG / |   Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 66 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 66 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.14 Å3/Da / Density % sol: 42.53 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 8 Details: PROTEIN AT 17 MG/ML IN 10 MILLIMOLAR TRIS-HCL PH 8.0, 100 MILLIMOLAR NACL, 3 MILLIMOLAR DTT. PROTEIN PREINCUBATED OVER NIGHT AT 293 K WITH 10 MILLIMOLAR 3-PHENYL-PROPIONATE, 3 MILLIMOLAR DTT ...Details: PROTEIN AT 17 MG/ML IN 10 MILLIMOLAR TRIS-HCL PH 8.0, 100 MILLIMOLAR NACL, 3 MILLIMOLAR DTT. PROTEIN PREINCUBATED OVER NIGHT AT 293 K WITH 10 MILLIMOLAR 3-PHENYL-PROPIONATE, 3 MILLIMOLAR DTT AND 1.5 MILLIMOLAR PLP. DROPS MADE FROM 1 MICROLITER PROTEIN AND 1 MICROLITER RESERVOIR (225 MILLIMOLAR MGCL2, 16-20 % (W/V) PEG 3350). INHIBITOR SOAKED INTO PREFORMED CRYSTALS FOR 15 DAYS AT 10 MILLIMOLAR. CRYO: RESERVOIR SOLUTION COMPLEMENTED WITH 20 % (V/V) GLYCEROL AND 10 MILLIMOLAR INHIBITOR |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY  / Beamline: 14.1 / Wavelength: 0.9184 Å / Beamline: 14.1 / Wavelength: 0.9184 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: May 23, 2017 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9184 Å / Relative weight: 1 |
| Reflection | Resolution: 2.54→47.78 Å / Num. obs: 24853 / % possible obs: 99.2 % / Redundancy: 7.2 % / Biso Wilson estimate: 57.6 Å2 / CC1/2: 0.997 / Rrim(I) all: 0.177 / Net I/σ(I): 9.6 |
| Reflection shell | Resolution: 2.54→2.7 Å / Redundancy: 7.2 % / Mean I/σ(I) obs: 1.3 / Num. unique obs: 27219 / CC1/2: 0.598 / Rrim(I) all: 1.49 / % possible all: 95.2 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2ABJ Resolution: 2.54→47.78 Å / Cor.coef. Fo:Fc: 0.943 / Cor.coef. Fo:Fc free: 0.923 / SU B: 32.167 / SU ML: 0.299 / SU R Cruickshank DPI: 0.2607 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.319 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES: WITH TLS ADDED.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 170.5 Å2 / Biso mean: 55.45 Å2 / Biso min: 30.27 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.54→47.78 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Ens-ID: 1 / Number: 11108 / Refine-ID: X-RAY DIFFRACTION / Type: interatomic distance / Rms dev position: 0.08 Å / Weight position: 0.05
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.545→2.611 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
