 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7npx: Thioredoxin glutathione reductase from Schistosoma mansoni in com... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7npx | ||||||
|---|---|---|---|---|---|---|---|
| Title | Thioredoxin glutathione reductase from Schistosoma mansoni in complex with 3-(3-Methoxyquinoxalin-2-yl)propanoic acid at 24 hours of soaking | ||||||
 Components Components | Thioredoxin glutathione reductase | ||||||
 Keywords Keywords | PROTEIN BINDING / fragment binding / redox enzyme / inhibitor / flavoreductase | ||||||
| Function / homology |  Function and homology information Function and homology informationglutathione-disulfide reductase (NADPH) activity / thioredoxin-disulfide reductase (NADPH) / thioredoxin-disulfide reductase (NADPH) activity / cell redox homeostasis / glutathione metabolic process / flavin adenine dinucleotide binding / cellular response to oxidative stress / mitochondrion / metal ion binding / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.7 Å | ||||||
 Authors Authors | Fata, F. / Silvestri, I. / Williams, D.L. / Angelucci, F. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: Acs Infect Dis. / Year: 2021 Title: Probing the Surface of a Parasite Drug Target Thioredoxin Glutathione Reductase Using Small Molecule Fragments. Authors: Fata, F. / Silvestri, I. / Ardini, M. / Ippoliti, R. / Di Leandro, L. / Demitri, N. / Polentarutti, M. / Di Matteo, A. / Lyu, H. / Thatcher, G.R.J. / Petukhov, P.A. / Williams, D.L. / Angelucci, F. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7npx.cif.gz | 471.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7npx.ent.gz | 389.1 KB | Display | PDB format |
| PDBx/mmJSON format | 7npx.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/np/7npxftp://data.pdbj.org/pub/pdb/validation_reports/np/7npx | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6zlbC  6zlpC  6zp3C  6zstC  7b02C  2v6oS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 65108.043 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Details: The structure is relative to the Sec597Cys mutant / Source: (gene. exp.)  |
|---|
-Non-polymers , 6 types, 147 molecules 
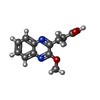




| #2: Chemical |  Mass: 785.550 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM Mass: 785.550 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM#3: Chemical |  Mass: 232.235 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H12N2O3 / Feature type: SUBJECT OF INVESTIGATION Mass: 232.235 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H12N2O3 / Feature type: SUBJECT OF INVESTIGATION#4: Chemical | ChemComp-CL /  Mass: 35.453 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Cl#5: Chemical | ChemComp-PEG /  Mass: 106.120 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 106.120 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C4H10O3#6: Chemical | ChemComp-K /  Mass: 39.098 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: K#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 131 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has ligand of interest | Y |
|---|---|
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.64 Å3/Da / Density % sol: 53.37 % |
|---|---|
| Crystal grow | Temperature: 294 K / Method: vapor diffusion, sitting drop / pH: 7.2 / Details: Bis-tris 0.1M PH = 7.0; Peg 3350 20%; KI 0.2 M |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ELETTRA  / Beamline: 5.2R / Wavelength: 1 Å / Beamline: 5.2R / Wavelength: 1 Å |
| Detector | Type: DECTRIS PILATUS 2M / Detector: PIXEL / Date: Jun 1, 2017 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.7→39.76 Å / Num. obs: 38527 / % possible obs: 99.8 % / Redundancy: 7.4 % / CC1/2: 0.998 / Net I/σ(I): 13.4 |
| Reflection shell | Resolution: 2.7→2.82 Å / Num. unique obs: 4577 / CC1/2: 0.787 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2V6O Resolution: 2.7→39.76 Å / Cor.coef. Fo:Fc: 0.938 / Cor.coef. Fo:Fc free: 0.914 / SU B: 30.208 / SU ML: 0.309 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.364 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : WITH TLS ADDED
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 156.41 Å2 / Biso mean: 63.477 Å2 / Biso min: 16.23 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.7→39.76 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.702→2.772 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: -16.808 Å / Origin y: -14.9778 Å / Origin z: 29.8622 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
