 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7fy1 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of apo human FABP9 | ||||||
 Components Components | Fatty acid-binding protein 9 | ||||||
 Keywords Keywords | LIPID BINDING PROTEIN / FATTY ACID BINDING PROTEIN / CYTOPLASM / LIPID-BINDING / TRANSPORT / PROTEIN BINDING | ||||||
| Function / homology |  Function and homology information Function and homology informationtriglyceride catabolic process / long-chain fatty acid binding / Triglyceride catabolism / lipid carrier activity / long-chain fatty acid transport / nucleus / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.01 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.01 Å | ||||||
 Authors Authors | Ehler, A. / Benz, J. / Obst, U. / Rudolph, M.G. | ||||||
| Funding support |  Switzerland, 1items Switzerland, 1items
| ||||||
 Citation Citation | Journal: Acta Crystallogr D Struct Biol / Year: 2025 Title: A high-resolution data set of fatty acid-binding protein structures. III. Unexpectedly high occurrence of wrong ligands. Authors: Ehler, A. / Bartelmus, C. / Benz, J. / Plitzko, I. / Rudolph, M.G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7fy1.cif.gz | 69.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7fy1.ent.gz | 51.4 KB | Display | PDB format |
| PDBx/mmJSON format | 7fy1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fy/7fy1ftp://data.pdbj.org/pub/pdb/validation_reports/fy/7fy1 | HTTPS FTP |
|---|
-Group deposition
| ID | G_1002264 (222 entries) |
|---|---|
| Title | To be published |
| Type | undefined |
| Description | A set of fabp crystal structures |










-Related structure data
| Similar structure data |
|---|
-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 15395.791 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: FABP9 / Plasmid: PET15b / Production host:  |
|---|---|
| #2: Chemical | ChemComp-MYR / 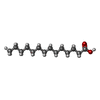  Mass: 228.371 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H28O2 Mass: 228.371 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H28O2 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 13 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 13 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | N |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.2 Å3/Da / Density % sol: 70.75 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 7 Details: protein in 25mM Tris/HCl pH 7.5 100mM NaCl, see also PMID 27658368 |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS / Beamline: X10SA / Wavelength: 1 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.01→46.76 Å / Num. obs: 18103 / % possible obs: 99.7 % / Redundancy: 35.999 % / Biso Wilson estimate: 50.64 Å2 / CC1/2: 1 / Rmerge(I) obs: 0.077 / Rrim(I) all: 0.078 / Χ2: 0.848 / Net I/σ(I): 23.84 / Num. measured all: 651695 / Scaling rejects: 55 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: inhouse model Resolution: 2.01→46.76 Å / SU ML: 0.24 / Cross valid method: THROUGHOUT / σ(F): 1.35 / Phase error: 32.89 / Stereochemistry target values: ML Details: loop with Phe56 at exit of ligand binding site has at least two conformations. mixture of fatty acids bound with one being shorter than the other, modeled by reducing occupancy of two ...Details: loop with Phe56 at exit of ligand binding site has at least two conformations. mixture of fatty acids bound with one being shorter than the other, modeled by reducing occupancy of two terminal CH2 to 0.5. data merged from two crystals since refines better than individual models.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.1 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 164.6 Å2 / Biso mean: 67.0738 Å2 / Biso min: 39.08 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.01→46.76 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / Total num. of bins used: 6
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
