 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7eup: Structural and mechanistic studies of a novel non-heme iron epime... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7eup | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structural and mechanistic studies of a novel non-heme iron epimerase/lyase and its utilization in chemoselective synthesis. | ||||||
 Components Components | Cupin domain-containing protein | ||||||
 Keywords Keywords | ISOMERASE / non-heme iron epimerase/lyase | ||||||
| Function / homology | RmlC-like cupin domain superfamily / RmlC-like jelly roll fold / metal ion binding / : / (2S,3R)-2-azanyl-3-phenyl-butanoic acid / Cupin domain-containing protein Function and homology information Function and homology information | ||||||
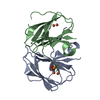
| Biological species |  Streptomyces albus (bacteria) Streptomyces albus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1104343131 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1104343131 Å | ||||||
 Authors Authors | Li, T.L. / Li, Y.S. / Chen, M.H. | ||||||
 Citation Citation | Journal: Acs Catalysis / Year: 2022 Title: Structural and Mechanistic Bases for StnK3 and Its Mutant-Mediated Lewis-Acid-Dependent Epimerization and Retro-Aldol Reactions. Authors: Chen, M.H. / Li, Y.S. / Hsu, N.S. / Lin, K.H. / Wang, Y.L. / Wang, Z.C. / Chang, C.F. / Lin, J.P. / Chang, C.Y. / Li, T.L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7eup.cif.gz | 71.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7eup.ent.gz | 42.3 KB | Display | PDB format |
| PDBx/mmJSON format | 7eup.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/eu/7eupftp://data.pdbj.org/pub/pdb/validation_reports/eu/7eup | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7eqkC  7eu6C  7eueC  7euzC  6j4cS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Ens-ID: 1 / End auth comp-ID: VAL / End label comp-ID: VAL
|
-Components
| #1: Protein | Mass: 13831.867 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Streptomyces albus (bacteria) / Gene: stnK3, G3260_000576 / Production host: #2: Chemical |   Mass: 55.845 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe / Feature type: SUBJECT OF INVESTIGATION Mass: 55.845 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe / Feature type: SUBJECT OF INVESTIGATION#3: Chemical | 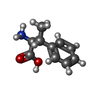  Mass: 179.216 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H13NO2 / Feature type: SUBJECT OF INVESTIGATION Mass: 179.216 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H13NO2 / Feature type: SUBJECT OF INVESTIGATION#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 69 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 69 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.71 Å3/Da / Density % sol: 54.64 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop Details: 1.2 M Sodium phosphate monobasic monohydrate, Potassium phosphate dibasic, pH 7.8 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSRRC  / Beamline: BL15A1 / Wavelength: 1 Å / Beamline: BL15A1 / Wavelength: 1 Å |
| Detector | Type: RAYONIX MX300HE / Detector: CCD / Date: Dec 5, 2020 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.11→30 Å / Num. obs: 17329 / % possible obs: 96.6 % / Redundancy: 7.5 % / Biso Wilson estimate: 31.4590347307 Å2 / Rsym value: 0.046 / Net I/σ(I): 25.7 |
| Reflection shell | Resolution: 2.11→2.19 Å / Num. unique obs: 1728 / Rsym value: 0.524 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 6J4C Resolution: 2.1104343131→23.1942902442 Å / SU ML: 0.188487912494 / Cross valid method: NONE / σ(F): 1.3677339183 / Phase error: 21.8877147141 Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 32.8330893076 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1104343131→23.1942902442 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
