 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7a11 | ||||||
|---|---|---|---|---|---|---|---|
| Title | LppS with covalent adduct derived from 1E | ||||||
 Components Components | L,D-transpeptidase 2 | ||||||
 Keywords Keywords | LIGASE / Transpeptidase / cell wall / peptidoglycan / antibiotic / Beta-lactam / Covalent inhibitor / Mycobacterium tuberculosis | ||||||
| Function / homology |  Function and homology information Function and homology informationpeptidoglycan-protein cross-linking / peptidoglycan L,D-transpeptidase activity / Transferases; Acyltransferases; Aminoacyltransferases / acyltransferase activity / cell wall organization / regulation of cell shape / metal ion binding / plasma membrane Similarity search - Function | ||||||
| Biological species |   Mycobacterium tuberculosis (bacteria) Mycobacterium tuberculosis (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.65 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.65 Å | ||||||
 Authors Authors | Schnell, R. / Steiner, E.M. | ||||||
| Funding support |  Sweden, 1items Sweden, 1items
| ||||||
 Citation Citation | Journal: Cell Chem Biol / Year: 2021 Title: N-Thio-beta-lactams targeting L,D-transpeptidase-2, with activity against drug-resistant strains of Mycobacterium tuberculosis. Authors: Martelli, G. / Pessatti, T.B. / Steiner, E.M. / Cirillo, M. / Caso, C. / Bisognin, F. / Landreh, M. / Monte, P.D. / Giacomini, D. / Schnell, R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7a11.cif.gz | 125.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7a11.ent.gz | 95.7 KB | Display | PDB format |
| PDBx/mmJSON format | 7a11.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/a1/7a11ftp://data.pdbj.org/pub/pdb/validation_reports/a1/7a11 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7a0zC  7a10C  7a1cC  7a1eC  5lbgS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 28219.289 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Details: The construct used here contains two domains: domain-B (Ig-like) and domain-C (catalytic transpeptidase domain), residue range 149-408 of the full length protein. Source: (gene. exp.) Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) (bacteria)Strain: ATCC 25618 / H37Rv / Gene: ldtB, lppS, Rv2518c, RVBD_2518c, P425_02624 / Plasmid: pNIC28-Bsa4 Details (production host): N-terminal His6-tag, removable by TEV cleavage Production host: References: UniProt: I6Y9J2, Transferases; Acyltransferases; Aminoacyltransferases #2: Chemical | ChemComp-XL3 / | 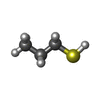  Mass: 76.161 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8S / Feature type: SUBJECT OF INVESTIGATION Mass: 76.161 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8S / Feature type: SUBJECT OF INVESTIGATION#3: Chemical | ChemComp-ACT /   Mass: 59.044 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 59.044 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C2H3O2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 537 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 537 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | Has protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.09 Å3/Da / Density % sol: 41.02 % / Description: rod |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 4.25 Details: Well solution: 0.1 M Na-Citrate pH 4.25 / 17.5% PEG 6K JM12 was added to the soaking solution at 4 mM concentration. In the soaking drop, Citrate is replaced by Acetate (0.3 M) 150 mM NaCl 0. ...Details: Well solution: 0.1 M Na-Citrate pH 4.25 / 17.5% PEG 6K JM12 was added to the soaking solution at 4 mM concentration. In the soaking drop, Citrate is replaced by Acetate (0.3 M) 150 mM NaCl 0.1 M Bis-Tris pH 6.2 0.3 M Na-Acetate pH 5.2 25% PEG 6K PEG 6K INCREASED to 25% FOR CRYO-PROTECTION |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: MAX IV / Beamline: BioMAX / Wavelength: 0.82656 Å |
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Mar 27, 2019 / Details: mirrors (VFM, HFM) |
| Radiation | Monochromator: Si(111) double crystal monochromator / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.82656 Å / Relative weight: 1 |
| Reflection | Resolution: 1.65→39.73 Å / Num. obs: 55802 / % possible obs: 100 % / Observed criterion σ(I): 1.8 / Redundancy: 7 % / Biso Wilson estimate: 21.6 Å2 / Rmerge(I) obs: 0.059 / Net I/σ(I): 14.2 |
| Reflection shell | Resolution: 1.65→1.68 Å / Rmerge(I) obs: 0.931 / Mean I/σ(I) obs: 1.8 / Num. unique obs: 2746 / % possible all: 100 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5LBG Resolution: 1.65→38 Å / Cor.coef. Fo:Fc: 0.973 / Cor.coef. Fo:Fc free: 0.955 / SU B: 2.261 / SU ML: 0.075 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.1 / ESU R Free: 0.102 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 84.86 Å2 / Biso mean: 27.62 Å2 / Biso min: 12.09 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.65→38 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.65→1.69 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
|
