 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6svb: Terahertz irradiated structure of bovine trypsin (odd frames of c... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6svb | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Terahertz irradiated structure of bovine trypsin (odd frames of crystal x40) | |||||||||
 Components Components | Cationic trypsin | |||||||||
 Keywords Keywords | HYDROLASE / bovine trypsin / Terahertz irradiation / odd frames / x40 | |||||||||
| Function / homology |  Function and homology information Function and homology informationtrypsin / serpin family protein binding / serine protease inhibitor complex / digestion / endopeptidase activity / serine-type endopeptidase activity / proteolysis / : / metal ion binding Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.15 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.15 Å | |||||||||
 Authors Authors | Ahlberg Gagner, V. / Lundholm, I. / Garcia-Bonete, M.J. / Rodilla, H. / Friedman, R. / Zhaunerchyk, V. / Bourenkov, G. / Schneider, T. / Stake, J. / Katona, G. | |||||||||
| Funding support |  Sweden, 2items Sweden, 2items
| |||||||||
 Citation Citation | Journal: Sci Rep / Year: 2019 Title: Clustering of atomic displacement parameters in bovine trypsin reveals a distributed lattice of atoms with shared chemical properties. Authors: Gagner, V.A. / Lundholm, I. / Garcia-Bonete, M.J. / Rodilla, H. / Friedman, R. / Zhaunerchyk, V. / Bourenkov, G. / Schneider, T. / Stake, J. / Katona, G. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6svb.cif.gz | 116.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6svb.ent.gz | 88.3 KB | Display | PDB format |
| PDBx/mmJSON format | 6svb.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/sv/6svbftp://data.pdbj.org/pub/pdb/validation_reports/sv/6svb | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6suxC  6sv0C  6sv6C  6sv8C  6sv9C  6svdC  6svgC  6sviC  6svjC  6svnC  6svrC  6svuC  6svvC  6svwC  6svxC  6svzC  6sw0C  418gS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |



















-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23324.287 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca | ||||||
| #3: Chemical | ChemComp-BEN / 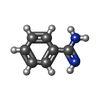  Mass: 120.152 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H8N2 Mass: 120.152 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H8N2 | ||||||
| #4: Chemical |   Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 288 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 288 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | N | Has protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.05 Å3/Da / Density % sol: 40.12 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 7 Details: protein solution - 60 mg/ml trypsin, 6 mg/ml benzamidine and 3 mM CaCl2 in a 30 mM HEPES buffer pH 7.0. Well solution - 18% PEG 8000, 200 mM (NH4)2SO4, 50 mM HEPES pH 7, 3 mM CaCl2 and 6 ...Details: protein solution - 60 mg/ml trypsin, 6 mg/ml benzamidine and 3 mM CaCl2 in a 30 mM HEPES buffer pH 7.0. Well solution - 18% PEG 8000, 200 mM (NH4)2SO4, 50 mM HEPES pH 7, 3 mM CaCl2 and 6 mg/ml benzamidine. 1:1 ratio drops |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: PETRA III, EMBL c/o DESY  / Beamline: P14 (MX2) / Wavelength: 0.8856 Å / Beamline: P14 (MX2) / Wavelength: 0.8856 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M-F / Detector: PIXEL / Date: Oct 23, 2015 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.8856 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.15→42.69 Å / Num. obs: 127574 / % possible obs: 95.6 % / Redundancy: 4.542 % / Biso Wilson estimate: 14.227 Å2 / CC1/2: 0.998 / Rmerge(I) obs: 0.105 / Rrim(I) all: 0.118 / Χ2: 1.142 / Net I/σ(I): 7.75 / Num. measured all: 579396 / Scaling rejects: 7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 418G Resolution: 1.15→42.69 Å / Cross valid method: FREE R-VALUE
| ||||||||||||||||
| Displacement parameters | Biso mean: 14.731 Å2 | ||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.15→42.69 Å
|
