| 登録情報 | データベース: PDB / ID: 6qsf
|
|---|
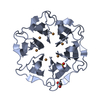
| タイトル | Crystal structure of Pizza6S in the presence of Keggin (STA) |
|---|
 要素 要素 | Pizza6S |
|---|
 キーワード キーワード | DE NOVO PROTEIN / Artificial beta-propeller protein / computational design / polyoxometalate / POM / Pizza / polyoxometalate-protein framework / POM-protein framework / Keggin structure / STA |
|---|
| 機能・相同性 | Keggin (STA) 機能・相同性情報 機能・相同性情報 |
|---|
| 生物種 | Synthetic construct (人工物) |
|---|
| 手法 |  X線回折 / シンクロトロン / 分子置換 / 解像度: 1.5 Å X線回折 / シンクロトロン / 分子置換 / 解像度: 1.5 Å |
|---|
 データ登録者 データ登録者 | Noguchi, H. / Vandebroek, L. / Kamata, K. / Tame, J.R.H. / Van Meervelt, L. / Parac-Vogt, T.N. / Voet, A.R.D. |
|---|
| 資金援助 |  ベルギー, ベルギー,  日本, 4件 日本, 4件 | 組織 | 認可番号 | 国 |
|---|
| G0E4717N | ベルギー | | G0F9316N | ベルギー | | G051917N | ベルギー | | 16H04779 | 日本 |
|
|---|
 引用 引用 | ジャーナル: Chem.Commun.(Camb.) / 年: 2020
タイトル: Hybrid assemblies of a symmetric designer protein and polyoxometalates with matching symmetry.
著者: Vandebroek, L. / Noguchi, H. / Kamata, K. / Tame, J.R.H. / Van Meervelt, L. / Parac-Vogt, T.N. / Voet, A.R.D. |
|---|
| 履歴 | | 登録 | 2019年2月20日 | 登録サイト: PDBE / 処理サイト: PDBE |
|---|
| 改定 1.0 | 2020年3月18日 | Provider: repository / タイプ: Initial release |
|---|
| 改定 1.1 | 2021年3月31日 | Group: Database references / カテゴリ: citation / citation_author
Item: _citation.country / _citation.journal_abbrev ..._citation.country / _citation.journal_abbrev / _citation.journal_id_CSD / _citation.journal_id_ISSN / _citation.journal_volume / _citation.page_first / _citation.page_last / _citation.pdbx_database_id_DOI / _citation.pdbx_database_id_PubMed / _citation.title / _citation.year / _citation_author.identifier_ORCID / _citation_author.name |
|---|
| 改定 1.2 | 2024年1月24日 | Group: Data collection / Database references / Refinement description
カテゴリ: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession |
|---|
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード













 PDBj
PDBj
 集合体
集合体



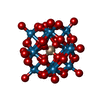
 分子量: 2874.142 Da / 分子数: 2 / 由来タイプ: 合成 / 式: O40SiW12 / タイプ: SUBJECT OF INVESTIGATION
分子量: 2874.142 Da / 分子数: 2 / 由来タイプ: 合成 / 式: O40SiW12 / タイプ: SUBJECT OF INVESTIGATION 分子量: 18.015 Da / 分子数: 233 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 233 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: I04 / 波長: 0.9795 Å
/ ビームライン: I04 / 波長: 0.9795 Å 解析
解析