 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6nyu: The X-ray crystal structure of Staphylococcus aureus Fatty Acid K... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6nyu | ||||||
|---|---|---|---|---|---|---|---|
| Title | The X-ray crystal structure of Staphylococcus aureus Fatty Acid Kinase (Fak) B1 F263T mutant protein to 2.18 Angstrom resolution | ||||||
 Components Components | DegV domain-containing protein | ||||||
 Keywords Keywords | TRANSFERASE / Fatty acid kinase | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |   Staphylococcus aureus (bacteria) Staphylococcus aureus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.183 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.183 Å | ||||||
 Authors Authors | Cuypers, M.G. / Gullett, J.M. / Subramanian, C. / White, S.W. / Rock, C.O. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: To Be Published Title: The X-ray crystal structure of STAPHYLOCOCCUS AUREUS Fatty Acid Kinase (Fak) B1 protein F263T mutant to 2.18 Angstrom resolution Authors: Cuypers, M.G. / Gullett, J.M. / Subramanian, C. / White, S.W. / Rock, C.O. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6nyu.cif.gz | 131.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6nyu.ent.gz | 100.3 KB | Display | PDB format |
| PDBx/mmJSON format | 6nyu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ny/6nyuftp://data.pdbj.org/pub/pdb/validation_reports/ny/6nyu | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5utoS S: Starting model for refinement |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj








- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 32055.436 Da / Num. of mol.: 2 / Mutation: F263T Source method: isolated from a genetically manipulated source Source: (gene. exp.) Staphylococcus aureus (bacteria)Gene: BTN44_08795, CV021_09110, EP54_02745, EQ90_03735, ERS072840_01626, HMPREF3211_01094, NCTC10654_00855, NCTC11940_00723, NCTC13131_00828, NCTC13196_00435, NCTC6133_00899, NCTC7878_03402, SAMEA1531701_00562 Production host: #2: Chemical | 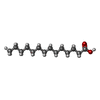  Mass: 228.371 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H28O2 / Feature type: SUBJECT OF INVESTIGATION Mass: 228.371 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H28O2 / Feature type: SUBJECT OF INVESTIGATION#3: Chemical |   Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3#4: Chemical |   Mass: 256.424 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C16H32O2 / Feature type: SUBJECT OF INVESTIGATION Mass: 256.424 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C16H32O2 / Feature type: SUBJECT OF INVESTIGATION#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 299 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 299 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.13 Å3/Da / Density % sol: 42.4 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop Details: PH 6.5 0.1M MES/IMIDAZOLE, 12.5% PEG1000, 12.5% PEG3350, 12.5% MPD, 0.03M NANO3, 0.03M NA2HPO4, 0.03M (NH4)2 SO4 Temp details: controlled room |
-Data collection
| Diffraction | Mean temperature: 100 K / Ambient temp details: cryostream - post 88K flash freezing / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 22-BM / Wavelength: 1 Å |
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD / Date: Dec 1, 2016 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.18→80.56 Å / Num. obs: 26506 / % possible obs: 96.6 % / Redundancy: 3.8 % / Biso Wilson estimate: 16.5 Å2 / CC1/2: 0.973 / Rmerge(I) obs: 0.134 / Rpim(I) all: 0.08 / Rrim(I) all: 0.156 / Net I/σ(I): 11.6 |
| Reflection shell | Resolution: 2.18→2.25 Å / Redundancy: 1.7 % / Rmerge(I) obs: 0.394 / Mean I/σ(I) obs: 3.8 / Num. unique obs: 1800 / CC1/2: 0.552 / Rpim(I) all: 0.379 / Rrim(I) all: 0.548 / % possible all: 76.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5UTO Resolution: 2.183→40.279 Å / SU ML: 0.25 / Cross valid method: FREE R-VALUE / σ(F): 1.99 / Phase error: 23.81
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.183→40.279 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
