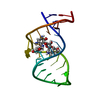
- PDB-6nk4: KVQIINKKL, crystal structure of a tau protein fragment -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 6nk4
Title
KVQIINKKL, crystal structure of a tau protein fragment
Components
Microtubule-associated protein tau
Keywords
STRUCTURAL PROTEIN / Amyloid / tau / Alzheimer's Disease / tauopathy / MAPT
Function / homology
Function and homology information
plus-end-directed organelle transport along microtubule / histone-dependent DNA binding / negative regulation of protein localization to mitochondrion / neurofibrillary tangle / microtubule lateral binding / axonal transport / tubulin complex / positive regulation of protein localization to synapse / phosphatidylinositol bisphosphate binding / generation of neurons ...plus-end-directed organelle transport along microtubule / histone-dependent DNA binding / negative regulation of protein localization to mitochondrion / neurofibrillary tangle / microtubule lateral binding / axonal transport / tubulin complex / positive regulation of protein localization to synapse / phosphatidylinositol bisphosphate binding / generation of neurons / rRNA metabolic process / axonal transport of mitochondrion / axon development / regulation of mitochondrial fission / regulation of microtubule-based movement / central nervous system neuron development / intracellular distribution of mitochondria / regulation of chromosome organization / minor groove of adenine-thymine-rich DNA binding / lipoprotein particle binding / microtubule polymerization / negative regulation of mitochondrial membrane potential / regulation of microtubule polymerization / dynactin binding / apolipoprotein binding / protein polymerization / main axon / Caspase-mediated cleavage of cytoskeletal proteins / regulation of microtubule polymerization or depolymerization / negative regulation of mitochondrial fission / axolemma / glial cell projection / neurofibrillary tangle assembly / positive regulation of axon extension / regulation of cellular response to heat / positive regulation of microtubule polymerization / positive regulation of protein localization / Activation of AMPK downstream of NMDARs / positive regulation of superoxide anion generation / cytoplasmic microtubule organization / cellular response to brain-derived neurotrophic factor stimulus / regulation of calcium-mediated signaling / regulation of long-term synaptic depression / supramolecular fiber organization / axon cytoplasm / somatodendritic compartment / synapse assembly / nuclear periphery / phosphatidylinositol binding / astrocyte activation / protein phosphatase 2A binding / enzyme inhibitor activity / stress granule assembly / regulation of autophagy / regulation of microtubule cytoskeleton organization / cellular response to reactive oxygen species / microglial cell activation / cellular response to nerve growth factor stimulus / Hsp90 protein binding / protein homooligomerization / SH3 domain binding / PKR-mediated signaling / regulation of synaptic plasticity / synapse organization / microtubule cytoskeleton organization / response to lead ion / memory / neuron projection development / cytoplasmic ribonucleoprotein granule / cell-cell signaling / cellular response to heat / single-stranded DNA binding / microtubule cytoskeleton / growth cone / protein-folding chaperone binding / actin binding / double-stranded DNA binding / cell body / microtubule binding / sequence-specific DNA binding / amyloid fibril formation / dendritic spine / microtubule / learning or memory / protein-macromolecule adaptor activity / neuron projection / membrane raft / negative regulation of gene expression / axon / neuronal cell body / DNA damage response / dendrite / protein kinase binding / enzyme binding / mitochondrion / DNA binding / RNA binding / extracellular region / identical protein binding / nucleus Similarity search - Function
Microtubule-associated protein Tau / Microtubule associated protein, tubulin-binding repeat / Tau and MAP protein, tubulin-binding repeat / Tau and MAP proteins tubulin-binding repeat signature. / Tau and MAP proteins tubulin-binding repeat profile. / : Similarity search - Domain/homology
ELECTRON CRYSTALLOGRAPHY / electron crystallography / MOLECULAR REPLACEMENT / cryo EM / Resolution: 1.994 Å
Model details
Crystal structure KVQIINKL from the microtubule binding region of tau protein reveals a tightly ...Crystal structure KVQIINKL from the microtubule binding region of tau protein reveals a tightly packed steric zipper
National Institutes of Health/National Institute on Aging (NIH/NIA)
AG0543022
United States
Citation
Journal: Proc Natl Acad Sci U S A / Year: 2021 Title: Intrinsic electronic conductivity of individual atomically resolved amyloid crystals reveals micrometer-long hole hopping via tyrosines. Authors: Catharine Shipps / H Ray Kelly / Peter J Dahl / Sophia M Yi / Dennis Vu / David Boyer / Calina Glynn / Michael R Sawaya / David Eisenberg / Victor S Batista / Nikhil S Malvankar / Abstract: Proteins are commonly known to transfer electrons over distances limited to a few nanometers. However, many biological processes require electron transport over far longer distances. For example, ...Proteins are commonly known to transfer electrons over distances limited to a few nanometers. However, many biological processes require electron transport over far longer distances. For example, soil and sediment bacteria transport electrons, over hundreds of micrometers to even centimeters, via putative filamentous proteins rich in aromatic residues. However, measurements of true protein conductivity have been hampered by artifacts due to large contact resistances between proteins and electrodes. Using individual amyloid protein crystals with atomic-resolution structures as a model system, we perform contact-free measurements of intrinsic electronic conductivity using a four-electrode approach. We find hole transport through micrometer-long stacked tyrosines at physiologically relevant potentials. Notably, the transport rate through tyrosines (10 s) is comparable to cytochromes. Our studies therefore show that amyloid proteins can efficiently transport charges, under ordinary thermal conditions, without any need for redox-active metal cofactors, large driving force, or photosensitizers to generate a high oxidation state for charge injection. By measuring conductivity as a function of molecular length, voltage, and temperature, while eliminating the dominant contribution of contact resistances, we show that a multistep hopping mechanism (composed of multiple tunneling steps), not single-step tunneling, explains the measured conductivity. Combined experimental and computational studies reveal that proton-coupled electron transfer confers conductivity; both the energetics of the proton acceptor, a neighboring glutamine, and its proximity to tyrosine influence the hole transport rate through a proton rocking mechanism. Surprisingly, conductivity increases 200-fold upon cooling due to higher availability of the proton acceptor by increased hydrogen bonding.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) MOLECULAR REPLACEMENT / cryo EM / Resolution: 1.994 Å
MOLECULAR REPLACEMENT / cryo EM / Resolution: 1.994 Å  Authors
Authors United States, 1items
United States, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads




 PDBj
PDBj





 Assembly
Assembly

 Mass: 18.015 Da / Num. of mol.: 3 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 3 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing