 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6nee: Crystal structure of a reconstructed ancestor of Triosephosphate ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6nee | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of a reconstructed ancestor of Triosephosphate isomerase from eukaryotes | ||||||
 Components Components | TRIOSEPHOSPHATE ISOMERASE | ||||||
 Keywords Keywords | ISOMERASE / Glycolisis / TIM Barrel / Ancestral sequence reconstruction | ||||||
| Function / homology | Aldolase class I / TIM Barrel / Alpha-Beta Barrel / Alpha Beta / PHOSPHOGLYCOLOHYDROXAMIC ACID Function and homology information Function and homology information | ||||||
| Biological species | synthetic construct (others) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Rodriguez-Romero, A. / Schulte-Sasse, M. / Fernandez-Velasco, D.A. | ||||||
 Citation Citation | Journal: FEBS J. / Year: 2019 Title: Structural, thermodynamic and catalytic characterization of an ancestral triosephosphate isomerase reveal early evolutionary coupling between monomer association and function. Authors: Schulte-Sasse, M. / Pardo-Avila, F. / Pulido-Mayoral, N.O. / Vazquez-Lobo, A. / Costas, M. / Garcia-Hernandez, E. / Rodriguez-Romero, A. / Fernandez-Velasco, D.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6nee.cif.gz | 118.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6nee.ent.gz | 90.8 KB | Display | PDB format |
| PDBx/mmJSON format | 6nee.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ne/6neeftp://data.pdbj.org/pub/pdb/validation_reports/ne/6nee | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2i9eS S: Starting model for refinement |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 27538.686 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) synthetic construct (others) / Plasmid: pET-28 b(+) / Production host:  #2: Chemical | 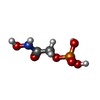  Mass: 171.046 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6NO6P Mass: 171.046 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6NO6P#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 427 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 427 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.32 Å3/Da / Density % sol: 46.97 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: 0.1M HEPS pH 7.5, 20% PEG 8000. Protein concentration 6 mg/mL in 10 mM Triethanolamine pH 7.6, 1 mM EDTA, 1 mM DTT, 50 mM NaCl, 10 mM PGH |
-Data collection
| Diffraction | Mean temperature: 103 K / Ambient temp details: oxford cryosystem / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 HF / Wavelength: 1.54 Å |
| Detector | Type: RIGAKU RAXIS IV++ / Detector: IMAGE PLATE / Date: Apr 29, 2016 / Details: Mirror |
| Radiation | Monochromator: Graphite monochromator / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→42.472 Å / Num. obs: 37060 / % possible obs: 95 % / Redundancy: 3.9 % / Biso Wilson estimate: 22.52 Å2 / CC1/2: 0.997 / Rmerge(I) obs: 0.071 / Net I/σ(I): 12.3 |
| Reflection shell | Resolution: 1.9→1.968 Å / Redundancy: 3.9 % / Rmerge(I) obs: 0.454 / Mean I/σ(I) obs: 3 / Num. unique obs: 2648 / % possible all: 91.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2I9E Resolution: 1.9→38.423 Å / SU ML: 0.2 / Cross valid method: THROUGHOUT / σ(F): 1.94 / Phase error: 22.15 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→38.423 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
