 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6kjm: Structural basis for domain rotation during adenylation of active... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6kjm | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structural basis for domain rotation during adenylation of active site K123 and fragment library screening against NAD+ -dependent DNA ligase from Mycobacterium tuberculosis | ||||||
 Components Components | DNA ligase A | ||||||
 Keywords Keywords | LIGASE | ||||||
| Function / homology |  Function and homology information Function and homology informationDNA ligase (NAD+) / DNA ligase (NAD+) activity / peptidoglycan-based cell wall / DNA replication / DNA repair / magnesium ion binding / plasma membrane / cytosol Similarity search - Function | ||||||
| Biological species |  Mycobacterium tuberculosis H37Rv (bacteria) Mycobacterium tuberculosis H37Rv (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å | ||||||
 Authors Authors | Ramachandran, R. / Shukla, A. / Afsar, M. | ||||||
 Citation Citation | Journal: J.Struct.Biol. / Year: 2021 Title: Structure based identification of first-in-class fragment inhibitors that target the NMN pocket of M. tuberculosis NAD + -dependent DNA ligase A. Authors: Shukla, A. / Afsar, M. / Kumar, N. / Kumar, S. / Ramachandran, R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6kjm.cif.gz | 133.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6kjm.ent.gz | 102.9 KB | Display | PDB format |
| PDBx/mmJSON format | 6kjm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/kj/6kjmftp://data.pdbj.org/pub/pdb/validation_reports/kj/6kjm | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6kkvC  6lw8C  1zauS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data | |
| Other databases |
|










-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 37607.883 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Mycobacterium tuberculosis H37Rv (bacteria)Strain: H37Rv / Gene: ligA / Production host: | ||||
|---|---|---|---|---|---|
| #2: Chemical | ChemComp-NMN / 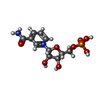  Mass: 335.227 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H16N2O8P / Feature type: SUBJECT OF INVESTIGATION Mass: 335.227 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H16N2O8P / Feature type: SUBJECT OF INVESTIGATION | ||||
| #3: Chemical | ChemComp-AMP /   Mass: 347.221 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H14N5O7P / Feature type: SUBJECT OF INVESTIGATION / Comment: AMP*YM Mass: 347.221 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H14N5O7P / Feature type: SUBJECT OF INVESTIGATION / Comment: AMP*YM | ||||
| #4: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 / Feature type: SUBJECT OF INVESTIGATION Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 / Feature type: SUBJECT OF INVESTIGATION#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 105 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 105 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.78 Å3/Da / Density % sol: 67.5 % |
|---|---|
| Crystal grow | Temperature: 298.15 K / Method: vapor diffusion, hanging drop / pH: 7.6 Details: 0.1M HEPES Na pH=7.6 0.1M NaCl 1.5M Ammonium Sulfate |
-Data collection
| Diffraction | Mean temperature: 100 K / Ambient temp details: cryo condition / Serial crystal experiment: N | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: RRCAT INDUS-2  / Beamline: PX-BL21 / Wavelength: 0.9795 Å / Beamline: PX-BL21 / Wavelength: 0.9795 Å | ||||||||||||||||||||||||||||||
| Detector | Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE / Date: Jan 24, 2019 | ||||||||||||||||||||||||||||||
| Radiation | Monochromator: Double crystal monochromator / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.9795 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.2→46.57 Å / Num. obs: 28579 / % possible obs: 100 % / Redundancy: 8 % / CC1/2: 0.998 / Rmerge(I) obs: 0.111 / Rpim(I) all: 0.041 / Rrim(I) all: 0.119 / Net I/σ(I): 15.9 / Num. measured all: 227755 / Scaling rejects: 2 | ||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1ZAU Resolution: 2.2→43.227 Å / SU ML: 0.24 / Cross valid method: THROUGHOUT / σ(F): 1.34 / Phase error: 23.4 Details: SF FILE CONTAINS FRIEDEL PAIRS UNDER I/F_MINUS AND I/F_PLUS COLUMNS.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 137.48 Å2 / Biso mean: 38.9705 Å2 / Biso min: 8.99 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.2→43.227 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / % reflection obs: 100 %
|
