| Entry | Database: PDB / ID: 6i33
|
|---|
| Title | Crystal structure of human glycine decarboxylase (P-protein) |
|---|
 Components Components | Glycine dehydrogenase (decarboxylating), mitochondrial |
|---|
 Keywords Keywords | OXIDOREDUCTASE / mitochondrial / glycine decarboxylase / glycine cleavage system P-protein |
|---|
| Function / homology |  Function and homology information Function and homology information
response to methylamine / response to lipoic acid / pyridoxal binding / glycine dehydrogenase (aminomethyl-transferring) / glycine catabolic process / glycine dehydrogenase (decarboxylating) activity / Glycine degradation / glycine decarboxylation via glycine cleavage system / glycine cleavage complex / glycine binding ...response to methylamine / response to lipoic acid / pyridoxal binding / glycine dehydrogenase (aminomethyl-transferring) / glycine catabolic process / glycine dehydrogenase (decarboxylating) activity / Glycine degradation / glycine decarboxylation via glycine cleavage system / glycine cleavage complex / glycine binding / cellular response to leukemia inhibitory factor / pyridoxal phosphate binding / electron transfer activity / lyase activity / mitochondrial matrix / protein homodimerization activity / mitochondrion / nucleoplasm / plasma membraneSimilarity search - Function Glycine dehydrogenase (decarboxylating) / Glycine cleavage system P protein / : / : / Glycine cleavage system P-protein / Glycine dehydrogenase, C-terminal domain / Aromatic amino acid beta-eliminating lyase/threonine aldolase / Beta-eliminating lyase / Pyridoxal phosphate-dependent transferase, small domain / Pyridoxal phosphate-dependent transferase, major domain / Pyridoxal phosphate-dependent transferaseSimilarity search - Domain/homology |
|---|
| Biological species |  Homo sapiens (human) Homo sapiens (human) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å |
|---|
 Authors Authors | Van Laer, B. / Kapp, U. / Leonard, G. / Mueller-Dieckmann, C. |
|---|
 Citation Citation | Journal: To Be Published
Title: Structural insights in human glycine decarboxylase and comparison with the Neanderthal variant
Authors: Van Laer, B. / Kapp, U. / Signor, L. / Leonard, G. / Mueller-Dieckmann, C. |
|---|
| History | | Deposition | Nov 5, 2018 | Deposition site: PDBE / Processing site: PDBE |
|---|
| Revision 1.0 | Nov 20, 2019 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Jan 15, 2020 | Group: Advisory / Derived calculations
Category: pdbx_unobs_or_zero_occ_atoms / pdbx_validate_close_contact / struct_conn |
|---|
| Revision 1.2 | Jan 24, 2024 | Group: Data collection / Database references ...Data collection / Database references / Derived calculations / Refinement description
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model / struct_conn / struct_ncs_dom_lim
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _struct_conn.pdbx_dist_value / _struct_ncs_dom_lim.beg_auth_comp_id / _struct_ncs_dom_lim.beg_label_asym_id / _struct_ncs_dom_lim.beg_label_comp_id / _struct_ncs_dom_lim.beg_label_seq_id / _struct_ncs_dom_lim.end_auth_comp_id / _struct_ncs_dom_lim.end_label_asym_id / _struct_ncs_dom_lim.end_label_comp_id / _struct_ncs_dom_lim.end_label_seq_id |
|---|
|
|---|
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads












 PDBj
PDBj


 Assembly
Assembly


 Mass: 247.142 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H10NO6P / Feature type: SUBJECT OF INVESTIGATION
Mass: 247.142 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H10NO6P / Feature type: SUBJECT OF INVESTIGATION
 Mass: 62.068 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: C2H6O2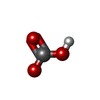
 Mass: 61.017 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CHO3 / Comment: pH buffer*YM
Mass: 61.017 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CHO3 / Comment: pH buffer*YM Mass: 18.015 Da / Num. of mol.: 165 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 165 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: MASSIF-3 / Wavelength: 0.97 Å
/ Beamline: MASSIF-3 / Wavelength: 0.97 Å Processing
Processing