Entry Database : PDB / ID : 6g4bTitle Crystal structure of the omega transaminase from Pseudomonas jessenii in the apo form, crystallized from succinate Aspartate aminotransferase family protein Keywords / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / Biological species Pseudomonas sp (bacteria)Method / / Resolution : 1.8 Å Authors Rozeboom, H.J. / Janssen, D.B. Journal : Febs J. / Year : 2019Title : Biochemical properties of a Pseudomonas aminotransferase involved in caprolactam metabolism.Authors : Palacio, C.M. / Rozeboom, H.J. / Lanfranchi, E. / Meng, Q. / Otzen, M. / Janssen, D.B. History Deposition Mar 27, 2018 Deposition site / Processing site Revision 1.0 Apr 10, 2019 Provider / Type Revision 1.1 Jun 19, 2019 Group / Database references / Category / citation_author / pdbx_database_procItem _citation.country / _citation.journal_abbrev ... _citation.country / _citation.journal_abbrev / _citation.journal_id_CSD / _citation.journal_id_ISSN / _citation.pdbx_database_id_DOI / _citation.pdbx_database_id_PubMed / _citation.title / _citation.year / _citation_author.name Revision 1.2 Oct 30, 2019 Group / Database references / Category / citation_authorItem _citation.journal_volume / _citation.page_first ... _citation.journal_volume / _citation.page_first / _citation.page_last / _citation_author.identifier_ORCID Revision 1.3 Jan 17, 2024 Group / Database references / Refinement descriptionCategory chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model / struct_ncs_dom_lim Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _struct_ncs_dom_lim.beg_auth_comp_id / _struct_ncs_dom_lim.beg_label_asym_id / _struct_ncs_dom_lim.beg_label_comp_id / _struct_ncs_dom_lim.beg_label_seq_id / _struct_ncs_dom_lim.end_auth_comp_id / _struct_ncs_dom_lim.end_label_asym_id / _struct_ncs_dom_lim.end_label_comp_id / _struct_ncs_dom_lim.end_label_seq_id
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Pseudomonas sp (bacteria)
Pseudomonas sp (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads














 PDBj
PDBj Assembly
Assembly
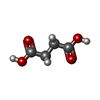
 Mass: 118.088 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H6O4
Mass: 118.088 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H6O4
 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 18.015 Da / Num. of mol.: 914 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 914 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing