 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6c63: Crystal Structure of the Mango-II Fluorescent Aptamer Bound to TO... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6c63 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of the Mango-II Fluorescent Aptamer Bound to TO1-Biotin | ||||||
 Components Components |
| ||||||
 Keywords Keywords | RNA / fluorescent / aptamer / G-quadruplex | ||||||
| Function / homology | Chem-EKJ / : / RNA / RNA (> 10) Function and homology information Function and homology information | ||||||
| Biological species | synthetic construct (others) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.90002801531 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.90002801531 Å | ||||||
 Authors Authors | Trachman, R.J. / Ferre-D'Amare, A.R. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2018 Title: Crystal Structures of the Mango-II RNA Aptamer Reveal Heterogeneous Fluorophore Binding and Guide Engineering of Variants with Improved Selectivity and Brightness. Authors: Trachman 3rd., R.J. / Abdolahzadeh, A. / Andreoni, A. / Cojocaru, R. / Knutson, J.R. / Ryckelynck, M. / Unrau, P.J. / Ferre-D'Amare, A.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6c63.cif.gz | 90.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6c63.ent.gz | 56.3 KB | Display | PDB format |
| PDBx/mmJSON format | 6c63.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 6c63_validation.pdf.gz | 1.2 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 6c63_full_validation.pdf.gz | 1.3 MB | Display | |
| Data in XML | 6c63_validation.xml.gz | 7 KB | Display | |
| Data in CIF | 6c63_validation.cif.gz | 8.7 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/c6/6c63ftp://data.pdbj.org/pub/pdb/validation_reports/c6/6c63 | HTTPS FTP |
-Related structure data
| Related structure data |  6c64C  6c65C 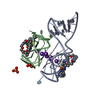 5v3fS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj





























- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 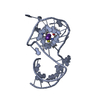
| ||||||||||||
| 2 | 
| ||||||||||||
| 3 | 
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: RNA chain | Mass: 11936.274 Da / Num. of mol.: 2 / Source method: obtained synthetically / Details: RNA was prepared by in vitro transcription / Source: (synth.) synthetic construct (others) #2: RNA chain | | Mass: 10635.501 Da / Num. of mol.: 1 / Source method: obtained synthetically / Details: RNA was prepared by in vitro transcription / Source: (synth.) synthetic construct (others) #3: Chemical | 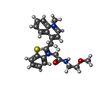  Mass: 407.529 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C23H25N3O2S Mass: 407.529 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C23H25N3O2S#4: Chemical | ChemComp-K /   Mass: 39.098 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: K |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.61 Å3/Da / Density % sol: 52.94 % |
|---|---|
| Crystal grow | Temperature: 294 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: 0.1 M HEPES pH 7.5, 2.45 M Ammonium Formate, 14.5% glycerol, 6.5% D-Sorbitol PH range: 7.0-7.5 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 24-ID-C / Wavelength: 1.495 Å / Beamline: 24-ID-C / Wavelength: 1.495 Å |
| Detector | Type: DECTRIS PILATUS3 6M / Detector: PIXEL / Date: Apr 7, 2017 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.495 Å / Relative weight: 1 |
| Reflection | Resolution: 2.9→91.206 Å / Num. obs: 15464 / % possible obs: 99 % / Redundancy: 8.8 % / Biso Wilson estimate: 74.48313766 Å2 / Rmerge(I) obs: 0.058 / Net I/σ(I): 4.4 |
| Reflection shell | Resolution: 2.9→2.99 Å / Redundancy: 8.2 % / Rmerge(I) obs: 0.34 / Num. unique obs: 1257 / % possible all: 99 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5V3F Resolution: 2.90002801531→91.206 Å / Rfactor Rfree error: 0.01 / SU ML: 0.30161926081 / Cross valid method: FREE R-VALUE / σ(F): 1.34361859768 / Phase error: 24.2447936435
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 76.7561539806 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.90002801531→91.206 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
