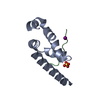
1.16 Angstrom Resolution Crystal Structure of Acyl Carrier Protein Domain (residues 1-100) of Polyketide Synthase Pks13 from Mycobacterium tuberculosis
Components
Polyketide synthase Pks13
Keywords
TRANSPORT PROTEIN / Structural Genomics / Center for Structural Genomics of Infectious Diseases / CSGID / Acyl Carrier Protein
Function / homology
Function and homology information
DIM/DIP cell wall layer assembly / fatty acid synthase activity / phosphopantetheine binding / 3-oxoacyl-[acyl-carrier-protein] synthase activity / Transferases; Acyltransferases; Transferring groups other than aminoacyl groups / fatty acid biosynthetic process / cytoplasm Similarity search - Function
Method to determine structure: SAD / Resolution: 1.16→26.13 Å / Cor.coef. Fo:Fc: 0.976 / Cor.coef. Fo:Fc free: 0.973 / SU B: 0.783 / SU ML: 0.016 / Cross valid method: THROUGHOUT / ESU R: 0.027 / ESU R Free: 0.027 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.14456
1793
5 %
RANDOM
Rwork
0.12952
-
-
-
obs
0.13029
34027
99.84 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 Mycobacterium tuberculosis (bacteria)
Mycobacterium tuberculosis (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads






 PDBj
PDBj


 Assembly
Assembly


 Mass: 118.174 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM
Mass: 118.174 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM
 Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 18.015 Da / Num. of mol.: 124 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 124 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 21-ID-F / Wavelength: 0.97872 Å
/ Beamline: 21-ID-F / Wavelength: 0.97872 Å Processing
Processing