 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6ap8: Crystal Structure of rice D14 bound to 2-(2-methyl-3-nitroanilino... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6ap8 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of rice D14 bound to 2-(2-methyl-3-nitroanilino)benzoic acid | |||||||||
 Components Components | Strigolactone esterase D14 | |||||||||
 Keywords Keywords | PLANT PROTEIN / alpha/beta hydrolase | |||||||||
| Function / homology |  Function and homology information Function and homology informationstrigolactone biosynthetic process / secondary shoot formation / Hydrolases; Acting on ester bonds / hydrolase activity / nucleus / cytoplasm Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.27 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.27 Å | |||||||||
 Authors Authors | Hamiaux, C. | |||||||||
| Funding support |  New Zealand, 2items New Zealand, 2items
| |||||||||
 Citation Citation | Journal: J. Biol. Chem. / Year: 2018 Title: Inhibition of strigolactone receptors byN-phenylanthranilic acid derivatives: Structural and functional insights. Authors: Hamiaux, C. / Drummond, R.S.M. / Luo, Z. / Lee, H.W. / Sharma, P. / Janssen, B.J. / Perry, N.B. / Denny, W.A. / Snowden, K.C. #1: Journal: Curr. Biol. / Year: 2012Title: DAD2 is an alpha/beta hydrolase likely to be involved in the perception of the plant branching hormone, strigolactone. Authors: Hamiaux, C. / Drummond, R.S. / Janssen, B.J. / Ledger, S.E. / Cooney, J.M. / Newcomb, R.D. / Snowden, K.C. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6ap8.cif.gz | 253.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6ap8.ent.gz | 203.2 KB | Display | PDB format |
| PDBx/mmJSON format | 6ap8.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ap/6ap8ftp://data.pdbj.org/pub/pdb/validation_reports/ap/6ap8 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6ap6C  6ap7C  3w04S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||
| 2 | 
| ||||||||||||||||||
| Unit cell |
| ||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: _ / Ens-ID: 1 / Beg auth comp-ID: GLY / Beg label comp-ID: GLY / End auth comp-ID: ARG / End label comp-ID: ARG / Refine code: _ / Auth seq-ID: 53 - 317 / Label seq-ID: 4 - 268
| ||||||||||||||||||
| Details | Monomer as determined by gel filtration |
-Components
| #1: Protein | Mass: 29366.695 Da / Num. of mol.: 2 / Fragment: residues 52-318 Source method: isolated from a genetically manipulated source Details: gene was codon optimized for E.coli expression Source: (gene. exp.) Gene: D14, D88, HTD2, Os03g0203200, LOC_Os03g10620 / Plasmid: pDEST566 / Production host:  References: UniProt: Q10QA5, Hydrolases; Acting on ester bonds #2: Chemical | 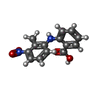  Mass: 272.256 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H12N2O4 / Feature type: SUBJECT OF INVESTIGATION Mass: 272.256 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H12N2O4 / Feature type: SUBJECT OF INVESTIGATION#3: Chemical |   Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 647 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 647 / Source method: isolated from a natural source / Formula: H2OHas protein modification | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.16 Å3/Da / Density % sol: 43.03 % / Description: Rod |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 7.5 / Details: HEPES 0.1M, MPD 5%, PEG 6000 8% |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Australian Synchrotron  / Beamline: MX2 / Wavelength: 0.9537 Å / Beamline: MX2 / Wavelength: 0.9537 Å | ||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 210r / Detector: CCD / Date: Nov 20, 2015 | ||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.9537 Å / Relative weight: 1 | ||||||||||||||||||||||||
| Reflection | Resolution: 1.27→48.01 Å / Num. obs: 134179 / % possible obs: 100 % / Redundancy: 14.5 % / Biso Wilson estimate: 10.3 Å2 / CC1/2: 0.999 / Rmerge(I) obs: 0.154 / Rpim(I) all: 0.042 / Rrim(I) all: 0.159 / Net I/σ(I): 12 | ||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR | Model details: Phaser MODE: MR_AUTO
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3W04 Resolution: 1.27→44.52 Å / Cor.coef. Fo:Fc: 0.983 / Cor.coef. Fo:Fc free: 0.972 / SU B: 1.573 / SU ML: 0.029 / SU R Cruickshank DPI: 0.0382 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.038 / ESU R Free: 0.04 Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.7 Å / Shrinkage radii: 0.7 Å / VDW probe radii: 1.2 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 77.57 Å2 / Biso mean: 13.154 Å2 / Biso min: 4.49 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.27→44.52 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Ens-ID: 1 / Number: 31812 / Refine-ID: X-RAY DIFFRACTION / Type: interatomic distance / Rms dev position: 0.13 Å / Weight position: 0.05
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.27→1.303 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
|
