Entry Database : PDB / ID : 5w71Title X-ray structure of BtrR from Bacillus circulans in the presence of the 2-DOS external aldimine L-glutamine:2-deoxy-scyllo-inosose aminotransferase Keywords / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / Biological species Bacillus circulans (bacteria)Method / / Resolution : 2.1 Å Authors Zachman-Brockmeyer, T.R. / Thoden, J.B. / Holden, H.M. Funding support Organization Grant number Country National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS) GM115921
Journal : Protein Sci. / Year : 2017Title : The structure of RbmB from Streptomyces ribosidificus, an aminotransferase involved in the biosynthesis of ribostamycin.Authors : Zachman-Brockmeyer, T.R. / Thoden, J.B. / Holden, H.M. History Deposition Jun 19, 2017 Deposition site / Processing site Revision 1.0 Jul 12, 2017 Provider / Type Revision 1.1 Jul 19, 2017 Group / Category Item _citation.country / _citation.journal_abbrev ... _citation.country / _citation.journal_abbrev / _citation.journal_id_ASTM / _citation.journal_id_CSD / _citation.journal_id_ISSN / _citation.pdbx_database_id_DOI / _citation.pdbx_database_id_PubMed / _citation.title / _citation.year Revision 1.2 Sep 6, 2017 Group / Category Item _citation.journal_volume / _citation.page_first ... _citation.journal_volume / _citation.page_first / _citation.page_last / _citation.title Revision 1.3 Sep 27, 2017 Group / Category / Item Revision 1.4 Jan 1, 2020 Group / Category / Item Revision 1.5 Oct 4, 2023 Group / Database references / Refinement descriptionCategory chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model Item / _database_2.pdbx_database_accession
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Bacillus circulans (bacteria)
Bacillus circulans (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United States, 1items
United States, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj Assembly
Assembly

 Mass: 247.142 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H10NO6P
Mass: 247.142 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H10NO6P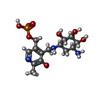
 Mass: 393.329 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H24N3O8P
Mass: 393.329 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H24N3O8P
 Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 18.015 Da / Num. of mol.: 384 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 384 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing