 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5v8c | ||||||
|---|---|---|---|---|---|---|---|
| Title | LytR-Csp2A-Psr enzyme from Actinomyces oris | ||||||
 Components Components | Transcriptional regulator | ||||||
 Keywords Keywords | TRANSCRIPTION / LCP / LytR-Cps2A-Psr / protein glycosylation / Gram positive surface glycosylation | ||||||
| Function / homology | Cell envelope-related transcriptional attenuator domain / LytR_cpsA_psr family / membrane => GO:0016020 / : / PHOSPHATE ION / Transcriptional regulator Function and homology information Function and homology information | ||||||
| Biological species |  Actinomyces oris (bacteria) Actinomyces oris (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.51 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.51 Å | ||||||
 Authors Authors | Amer, B.R. / Sawaya, M.R. / Liauw, B. / Fu, J.Y. / Ton-That, H. / Clubb, R.T. | ||||||
 Citation Citation | Journal: MBio / Year: 2019 Title: Structure and Mechanism of LcpA, a Phosphotransferase That Mediates Glycosylation of a Gram-Positive Bacterial Cell Wall-Anchored Protein. Authors: Siegel, S.D. / Amer, B.R. / Wu, C. / Sawaya, M.R. / Gosschalk, J.E. / Clubb, R.T. / Ton-That, H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5v8c.cif.gz | 113.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5v8c.ent.gz | 85 KB | Display | PDB format |
| PDBx/mmJSON format | 5v8c.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/v8/5v8cftp://data.pdbj.org/pub/pdb/validation_reports/v8/5v8c | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 30964.828 Da / Num. of mol.: 1 / Fragment: UNP residues 71-363 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Actinomyces oris (bacteria) / Gene: AXE84_08820 / Plasmid: pSUMO / Production host: |
|---|---|
| #2: Chemical | ChemComp-CO /   Mass: 58.933 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Co Mass: 58.933 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Co |
| #3: Chemical | ChemComp-PO4 /   Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 |
| #4: Chemical | ChemComp-PE4 / 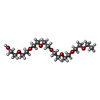  Mass: 354.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H34O8 / Comment: precipitant*YM Mass: 354.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H34O8 / Comment: precipitant*YM |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 6 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 6 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.87 Å3/Da / Density % sol: 34.16 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 5.5 Details: 0.1M sodium citrate pH 5.5, 25% PEG4000, 20% 2-propanol, 5 mM DTT, 200 mM NaCl, 5 mM MgCl2 |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 24-ID-C / Wavelength: 0.979 Å / Beamline: 24-ID-C / Wavelength: 0.979 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M-F / Detector: PIXEL / Date: Nov 6, 2015 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.979 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.51→52.83 Å / Num. obs: 8332 / % possible obs: 98.9 % / Observed criterion σ(I): -3 / Redundancy: 6.354 % / Biso Wilson estimate: 89.27 Å2 / CC1/2: 0.998 / Rmerge(I) obs: 0.051 / Rrim(I) all: 0.056 / Χ2: 0.997 / Net I/σ(I): 16.37 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.51→52.83 Å / Cor.coef. Fo:Fc: 0.95 / Cor.coef. Fo:Fc free: 0.923 / SU R Cruickshank DPI: 1.381 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 2.418 / SU Rfree Blow DPI: 0.333 / SU Rfree Cruickshank DPI: 0.335
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 188.51 Å2 / Biso mean: 98.01 Å2 / Biso min: 63.88 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.37 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.51→52.83 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.51→2.81 Å / Rfactor Rfree error: 0 / Total num. of bins used: 5
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 10.6784 Å / Origin y: 4.3011 Å / Origin z: 7.7936 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: { A|78 - A|368 } |
