| Entry | Database: PDB / ID: 5o0z
|
|---|
| Title | Structure of laspartomycin C in complex with geranyl-phosphate |
|---|
 Components Components | Laspartomycin C |
|---|
 Keywords Keywords | ANTIBIOTIC / antibiotic complex |
|---|
| Function / homology | : / [(~{E})-3-methylhex-2-enyl] dihydrogen phosphate / ACETIC ACID Function and homology information Function and homology information |
|---|
| Biological species |  Streptomyces viridochromogenes (bacteria) Streptomyces viridochromogenes (bacteria) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / SIRAS / Resolution: 1.28 Å X-RAY DIFFRACTION / SYNCHROTRON / SIRAS / Resolution: 1.28 Å |
|---|
 Authors Authors | Vlieg, H.C. / Kleijn, L.H.J. / Martin, N.I. / Janssen, B.J.C. |
|---|
| Funding support |  Netherlands, 1items Netherlands, 1items | Organization | Grant number | Country |
|---|
| | Netherlands |
|
|---|
 Citation Citation | Journal: Angew. Chem. Int. Ed. Engl. / Year: 2017
Title: A High-Resolution Crystal Structure that Reveals Molecular Details of Target Recognition by the Calcium-Dependent Lipopeptide Antibiotic Laspartomycin C.
Authors: Kleijn, L.H.J. / Vlieg, H.C. / Wood, T.M. / Sastre Torano, J. / Janssen, B.J.C. / Martin, N.I. |
|---|
| History | | Deposition | May 17, 2017 | Deposition site: PDBE / Processing site: PDBE |
|---|
| Revision 1.0 | Nov 15, 2017 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Dec 13, 2017 | Group: Database references / Category: citation / Item: _citation.title |
|---|
| Revision 1.2 | Jan 3, 2018 | Group: Database references / Category: citation
Item: _citation.journal_volume / _citation.page_first / _citation.page_last |
|---|
| Revision 1.3 | Oct 16, 2019 | Group: Data collection / Category: reflns_shell |
|---|
| Revision 2.0 | Aug 7, 2024 | Group: Advisory / Atomic model ...Advisory / Atomic model / Data collection / Database references / Derived calculations / Structure summary
Category: atom_site / atom_site_anisotrop ...atom_site / atom_site_anisotrop / chem_comp_atom / chem_comp_bond / database_2 / pdbx_molecule_features / pdbx_poly_seq_scheme / pdbx_struct_conn_angle / pdbx_unobs_or_zero_occ_atoms / struct_conn / struct_ref_seq / struct_site / struct_site_gen
Item: _atom_site.auth_seq_id / _atom_site_anisotrop.pdbx_auth_seq_id ..._atom_site.auth_seq_id / _atom_site_anisotrop.pdbx_auth_seq_id / _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_poly_seq_scheme.pdb_seq_num / _pdbx_struct_conn_angle.ptnr1_auth_asym_id / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr3_auth_asym_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _pdbx_unobs_or_zero_occ_atoms.auth_seq_id / _struct_conn.conn_type_id / _struct_conn.id / _struct_conn.pdbx_dist_value / _struct_conn.pdbx_leaving_atom_flag / _struct_conn.ptnr1_auth_asym_id / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_asym_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id / _struct_ref_seq.db_align_beg / _struct_ref_seq.db_align_end / _struct_ref_seq.pdbx_auth_seq_align_beg / _struct_ref_seq.pdbx_auth_seq_align_end / _struct_site.pdbx_auth_seq_id / _struct_site_gen.auth_seq_id |
|---|
|
|---|
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads




 PDBj
PDBj

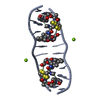
 Assembly
Assembly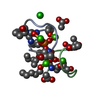

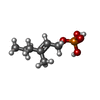




 Mass: 194.165 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C7H15O4P
Mass: 194.165 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C7H15O4P Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca
Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 60.052 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H4O2
Mass: 60.052 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H4O2 Sample preparation
Sample preparation / Beamline: ID23-2 / Wavelength: 0.873 Å
/ Beamline: ID23-2 / Wavelength: 0.873 Å Processing
Processing