- PDB-5jd8: Crystal structure of the serine endoprotease from Yersinia pestis -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 5jd8
Title
Crystal structure of the serine endoprotease from Yersinia pestis
Components
Periplasmic serine peptidase DegS
Keywords
HYDROLASE / DegS / Structural Genomics / Center for Structural Genomics of Infectious Diseases / CSGID
Function / homology
Function and homology information
protein quality control for misfolded or incompletely synthesized proteins / Hydrolases; Acting on peptide bonds (peptidases); Serine endopeptidases / periplasmic space / serine-type endopeptidase activity Similarity search - Function
Resolution: 1.85→30 Å / Cor.coef. Fo:Fc: 0.966 / Cor.coef. Fo:Fc free: 0.946 / SU B: 4.311 / SU ML: 0.064 / Cross valid method: THROUGHOUT / ESU R: 0.103 / ESU R Free: 0.102 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.18269
1292
4.9 %
RANDOM
Rwork
0.14831
-
-
-
obs
0.14999
25187
99.91 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Yersinia pestis CO92 (bacteria)
Yersinia pestis CO92 (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads






 PDBj
PDBj
 Assembly
Assembly

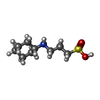
 Mass: 221.317 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H19NO3S / Comment: pH buffer*YM
Mass: 221.317 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H19NO3S / Comment: pH buffer*YM
 Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3
Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3
 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 18.015 Da / Num. of mol.: 154 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 154 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 21-ID-F / Wavelength: 0.97872 Å
/ Beamline: 21-ID-F / Wavelength: 0.97872 Å Processing
Processing