 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4xdg: Crystal Structure of Quinone Reductase II in complex with 2-(4-am... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4xdg | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Quinone Reductase II in complex with 2-(4-aminophenyl)-5-methoxy-1-oxy-indol-3-one molecule | ||||||
 Components Components | Ribosyldihydronicotinamide dehydrogenase [quinone] | ||||||
 Keywords Keywords | OXIDOREDUCTASE / QR2 / FAD / FLAVOPROTEIN / METAL-BINDING / indolone oxide | ||||||
| Function / homology |  Function and homology information Function and homology informationribosyldihydronicotinamide dehydrogenase (quinone) / dihydronicotinamide riboside quinone reductase activity / quinone catabolic process / resveratrol binding / oxidoreductase activity, acting on other nitrogenous compounds as donors / melatonin binding / NAD(P)H dehydrogenase (quinone) activity / Phase I - Functionalization of compounds / chloride ion binding / FAD binding ...ribosyldihydronicotinamide dehydrogenase (quinone) / dihydronicotinamide riboside quinone reductase activity / quinone catabolic process / resveratrol binding / oxidoreductase activity, acting on other nitrogenous compounds as donors / melatonin binding / NAD(P)H dehydrogenase (quinone) activity / Phase I - Functionalization of compounds / chloride ion binding / FAD binding / electron transfer activity / oxidoreductase activity / protein homodimerization activity / extracellular exosome / zinc ion binding / nucleoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å | ||||||
 Authors Authors | Sirigu, S. / Nepveu, F. / Vuillard, L. / Ferry, G. / Isabet, T. / Thompson, A. / Boutin, J.A. | ||||||
 Citation Citation | Journal: Molecules / Year: 2017 Title: Role of Quinone Reductase 2 in the Antimalarial Properties of Indolone-Type Derivatives. Authors: Cassagnes, L.E. / Rakotoarivelo, N. / Sirigu, S. / Perio, P. / Najahi, E. / Chavas, L.M. / Thompson, A. / Gayon, R. / Ferry, G. / Boutin, J.A. / Valentin, A. / Reybier, K. / Nepveu, F. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4xdg.cif.gz | 216.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4xdg.ent.gz | 171.2 KB | Display | PDB format |
| PDBx/mmJSON format | 4xdg.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 4xdg_validation.pdf.gz | 1.4 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 4xdg_full_validation.pdf.gz | 1.4 MB | Display | |
| Data in XML | 4xdg_validation.xml.gz | 24.8 KB | Display | |
| Data in CIF | 4xdg_validation.cif.gz | 37.3 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xd/4xdgftp://data.pdbj.org/pub/pdb/validation_reports/xd/4xdg | HTTPS FTP |
-Related structure data











-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 25980.533 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: NQO2, NMOR2 / Production host:  |
|---|
-Non-polymers , 5 types, 533 molecules 

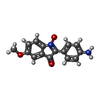


| #2: Chemical |  Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#3: Chemical |  Mass: 785.550 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM Mass: 785.550 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM#4: Chemical |  Mass: 268.267 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H12N2O3 Mass: 268.267 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H12N2O3#5: Chemical |  Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 524 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.42 Å3/Da / Density % sol: 49.12 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 7.5 / Details: 1.4M Ammonium Sulphate; 100 mM Hepes pH7.5 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SOLEIL  / Beamline: PROXIMA 1 / Wavelength: 0.97857 Å / Beamline: PROXIMA 1 / Wavelength: 0.97857 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Sep 19, 2014 |
| Radiation | Monochromator: chanel cut Si III / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97857 Å / Relative weight: 1 |
| Reflection | Resolution: 1.5→50 Å / Num. obs: 81302 / % possible obs: 99.9 % / Redundancy: 7.31 % / Biso Wilson estimate: 17.62 Å2 / Rsym value: 0.009 / Net I/σ(I): 12.58 |
| Reflection shell | Resolution: 1.5→1.59 Å / Redundancy: 7.14 % / Mean I/σ(I) obs: 3.11 / % possible all: 99.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.5→46.81 Å / Cor.coef. Fo:Fc: 0.9631 / Cor.coef. Fo:Fc free: 0.9592 / SU R Cruickshank DPI: 0.066 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.069 / SU Rfree Blow DPI: 0.068 / SU Rfree Cruickshank DPI: 0.065
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 26.25 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.217 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 1.5→46.81 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.5→1.54 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
