| Entry | Database: PDB / ID: 4tmc
|
|---|
| Title | CRYSTAL STRUCTURE of OLD YELLOW ENZYME from CANDIDA MACEDONIENSIS AKU4588 COMPLEXED with P-HYDROXYBENZALDEHYDE |
|---|
 Components Components | Old yellow enzyme |
|---|
 Keywords Keywords | FLAVOPROTEIN / TIM BARREL MOTIF / DEHYDROGENASE |
|---|
| Function / homology |  Function and homology information Function and homology information
Oxidoreductase Oye-like / NADH:flavin oxidoreductase/NADH oxidase, N-terminal / NADH:flavin oxidoreductase / NADH oxidase family / Aldolase class I / Aldolase-type TIM barrel / TIM Barrel / Alpha-Beta Barrel / Alpha BetaSimilarity search - Domain/homology |
|---|
| Biological species |  Kluyveromyces marxianus (yeast) Kluyveromyces marxianus (yeast) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å |
|---|
 Authors Authors | Horita, S. / Kataoka, M. / Kitamura, N. / Nakagawa, T. / Miyakawa, T. / Ohtsuka, J. / Nagata, K. / Shimizu, S. / Tanokura, M. |
|---|
| Funding support |  Japan, 4items Japan, 4items | Organization | Grant number | Country |
|---|
| the National Project on Protein Structural and Functional Analyses | | Japan | | the Targeted Proteins Research Program | | Japan | | the Platform for Drug Discovery, Informatics, and Structural Life Sciences | | Japan | | Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan | | Japan |
|
|---|
 Citation Citation | Journal: Chembiochem / Year: 2015
Title: An Engineered Old Yellow Enzyme that Enables Efficient Synthesis of (4R,6R)-Actinol in a One-Pot Reduction System
Authors: Horita, S. / Kataoka, M. / Kitamura, N. / Nakagawa, T. / Miyakawa, T. / Ohtsuka, J. / Nagata, K. / Shimizu, S. / Tanokura, M. |
|---|
| History | | Deposition | May 31, 2014 | Deposition site: RCSB / Processing site: PDBJ |
|---|
| Revision 1.0 | Feb 11, 2015 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Jan 29, 2020 | Group: Data collection / Derived calculations / Source and taxonomy
Category: diffrn_source / entity_src_gen / pdbx_struct_oper_list
Item: _diffrn_source.pdbx_synchrotron_site / _entity_src_gen.pdbx_alt_source_flag / _pdbx_struct_oper_list.symmetry_operation |
|---|
| Revision 1.2 | Nov 8, 2023 | Group: Data collection / Database references / Refinement description
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / diffrn_radiation_wavelength / pdbx_initial_refinement_model
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession |
|---|
|
|---|
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj Assembly
Assembly



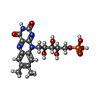
 Mass: 456.344 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C17H21N4O9P
Mass: 456.344 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C17H21N4O9P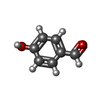
 Mass: 122.121 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C7H6O2
Mass: 122.121 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C7H6O2 Mass: 18.015 Da / Num. of mol.: 1050 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 1050 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing