 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4haq: Crystal Structure of a GH7 family cellobiohydrolase from Limnoria... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4haq | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of a GH7 family cellobiohydrolase from Limnoria quadripunctata in complex with cellobiose and cellotriose | ||||||||||||
 Components Components | GH7 family protein | ||||||||||||
 Keywords Keywords | HYDROLASE / cellobiohydrolase | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationcellulose 1,4-beta-cellobiosidase (non-reducing end) / cellulose 1,4-beta-cellobiosidase activity / cellulose catabolic process / extracellular region Similarity search - Function | ||||||||||||
| Biological species |  Limnoria quadripunctata (arthropod) Limnoria quadripunctata (arthropod) | ||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.9 Å | ||||||||||||
 Authors Authors | Martin, R.N.A. / McGeehan, J.E. / Streeter, S.D. / Cragg, S.M. / Guille, M.J. / Schnorr, K.M. / Kern, M. / Bruce, N.C. / McQueen-Mason, S.J. | ||||||||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2013 Title: Structural characterization of a unique marine animal family 7 cellobiohydrolase suggests a mechanism of cellulase salt tolerance Authors: Kern, M. / McGeehan, J.E. / Streeter, S.D. / Martin, R.N. / Besser, K. / Elias, L. / Eborall, W. / Malyon, G.P. / Payne, C.M. / Himmel, M.E. / Schnorr, K. / Beckham, G.T. / Cragg, S.M. / ...Authors: Kern, M. / McGeehan, J.E. / Streeter, S.D. / Martin, R.N. / Besser, K. / Elias, L. / Eborall, W. / Malyon, G.P. / Payne, C.M. / Himmel, M.E. / Schnorr, K. / Beckham, G.T. / Cragg, S.M. / Bruce, N.C. / McQueen-Mason, S.J. #1: Journal: Proc.Natl.Acad.Sci.USA / Year: 2010 Title: Molecular insight into lignocellulose digestion by a marine isopod in the absence of gut microbes. Authors: King, A.J. / Cragg, S.M. / Li, Y. / Dymond, J. / Guille, M.J. / Bowles, D.J. / Bruce, N.C. / Graham, I.A. / McQueen-Mason, S.J. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4haq.cif.gz | 191.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4haq.ent.gz | 147.5 KB | Display | PDB format |
| PDBx/mmJSON format | 4haq.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ha/4haqftp://data.pdbj.org/pub/pdb/validation_reports/ha/4haq | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4gwaSC  4hapC  4ipmC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 46535.949 Da / Num. of mol.: 2 / Fragment: UNP RESIDUES 19-448 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Limnoria quadripunctata (arthropod) / Gene: GH7B / Production host:  References: UniProt: D4HRL0, cellulose 1,4-beta-cellobiosidase (non-reducing end) |
|---|
-Sugars , 2 types, 3 molecules
| #2: Polysaccharide |   Source method: isolated from a genetically manipulated source Details: oligosaccharide / References: beta-cellobiose #3: Polysaccharide | beta-D-glucopyranose-(1-4)-beta-D-glucopyranose-(1-4)-alpha-D-glucopyranose / alpha-cellotriose | 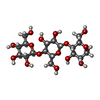  Source method: isolated from a genetically manipulated source Details: oligosaccharide / References: alpha-cellotriose |
|---|
-Non-polymers , 4 types, 568 molecules 



| #4: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg | ||
|---|---|---|---|
| #5: Chemical | ChemComp-TRS /  Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM | ||
| #6: Chemical |  Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 564 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.19 Å3/Da / Density % sol: 43.83 % / Mosaicity: 0.64 ° |
|---|---|
| Crystal grow | Temperature: 289 K / Method: vapor diffusion / pH: 8 Details: 0.2M MAGNESIUM CHLORIDE, 0.1M BIS-TRIS, 25% w/v PEG 3350, PH 8.0, VAPOUR DIFFUSION, SITTING DROP, TEMPERATURE 289K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I02 / Wavelength: 0.9173 Å / Beamline: I02 / Wavelength: 0.9173 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: May 6, 2012 |
| Radiation | Monochromator: SI(111)DUAL CRYSTAL MONOCHROMATOR / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9173 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→45.04 Å / Num. obs: 59787 / % possible obs: 96 % / Redundancy: 3.4 % / Rsym value: 0.146 / Net I/σ(I): 6.7 |
| Reflection shell | Resolution: 1.9→2 Å / Redundancy: 3.5 % / Rmerge(I) obs: 0.513 / Mean I/σ(I) obs: 1.3 / Rsym value: 0.513 / % possible all: 95.4 |
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR | Model details: Phaser MODE: MR_AUTO
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4GWA Resolution: 1.9→44.62 Å / Cor.coef. Fo:Fc: 0.949 / Cor.coef. Fo:Fc free: 0.916 / WRfactor Rfree: 0.2062 / WRfactor Rwork: 0.1569 / Occupancy max: 1 / Occupancy min: 0.5 / FOM work R set: 0.8883 / SU B: 3.327 / SU ML: 0.098 / SU R Cruickshank DPI: 0.1597 / SU Rfree: 0.1475 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.165 / ESU R Free: 0.151 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN USED IF PRESENT IN THE INPUT U VALUES
| |||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 92.18 Å2 / Biso mean: 13.627 Å2 / Biso min: 3.11 Å2
| |||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→44.62 Å
| |||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→1.949 Å / Total num. of bins used: 20
|
