 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3t9z | ||||||
|---|---|---|---|---|---|---|---|
| Title | A. fulgidus GlnK3, ligand-free | ||||||
 Components Components | Nitrogen regulatory protein P-II (GlnB-3) | ||||||
 Keywords Keywords | SIGNALING PROTEIN / PII-family / regulator / Amt3 | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of nitrogen utilization / enzyme regulator activity / ATP binding / cytosol Similarity search - Function | ||||||
| Biological species |   Archaeoglobus fulgidus (archaea) Archaeoglobus fulgidus (archaea) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.82 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.82 Å | ||||||
 Authors Authors | Maier, S. / Schleberger, P. / Lue, W. / Wacker, T. / Pflueger, T. / Litz, C. / Andrade, S.L.A. | ||||||
 Citation Citation | Journal: Plos One / Year: 2011 Title: Mechanism of disruption of the Amt-GlnK complex by P(II)-mediated sensing of 2-oxoglutarate. Authors: Maier, S. / Schleberger, P. / Lu, W. / Wacker, T. / Pfluger, T. / Litz, C. / Andrade, S.L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3t9z.cif.gz | 126.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3t9z.ent.gz | 98.3 KB | Display | PDB format |
| PDBx/mmJSON format | 3t9z.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 3t9z_validation.pdf.gz | 484.6 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 3t9z_full_validation.pdf.gz | 488 KB | Display | |
| Data in XML | 3t9z_validation.xml.gz | 25.2 KB | Display | |
| Data in CIF | 3t9z_validation.cif.gz | 36.4 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/t9/3t9zftp://data.pdbj.org/pub/pdb/validation_reports/t9/3t9z | HTTPS FTP |
-Related structure data
| Related structure data |  3ta0C  3ta1C  3ta2C  3o8wS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 13164.234 Da / Num. of mol.: 6 / Fragment: UNP residues 12-120 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Archaeoglobus fulgidus (archaea)Strain: ATCC 49558 / VC-16 / DSM 4304 / JCM 9628 / NBRC 100126 Gene: AF_1750, glnK3 / Production host:  #2: Chemical | ChemComp-FLC / | 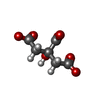  Mass: 189.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H5O7 Mass: 189.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H5O7#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 353 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 353 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.32 Å3/Da / Density % sol: 46.97 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 3.5 Details: 17% PEG3350, 0.1 M sodium citrate buffer, pH 3.5, VAPOR DIFFUSION, SITTING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06DA / Wavelength: 1 Å / Beamline: X06DA / Wavelength: 1 Å |
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD |
| Radiation | Monochromator: Bartels / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.82→64.4 Å / Num. all: 64689 / Num. obs: 63525 / % possible obs: 98.2 % / Observed criterion σ(F): 2 / Observed criterion σ(I): 2 / Redundancy: 3 % / Biso Wilson estimate: 29.29 Å2 / Rsym value: 0.048 / Net I/σ(I): 11 |
| Reflection shell | Resolution: 1.82→1.92 Å / Rmerge(I) obs: 0.191 / Mean I/σ(I) obs: 3.1 / Rsym value: 0.288 / % possible all: 98.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3O8W Resolution: 1.82→26.65 Å / Cor.coef. Fo:Fc: 0.9218 / Cor.coef. Fo:Fc free: 0.9047 / SU R Cruickshank DPI: 0.123 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.124 / SU Rfree Blow DPI: 0.117 / SU Rfree Cruickshank DPI: 0.117 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 40.66 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.246 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.82→26.65 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.82→1.87 Å / Total num. of bins used: 20
|
