 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3ssa: Crystal structure of subunit B mutant N157T of the A1AO ATP synthase -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3ssa | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of subunit B mutant N157T of the A1AO ATP synthase | ||||||
 Components Components | V-type ATP synthase beta chain | ||||||
 Keywords Keywords | HYDROLASE / ATP binding | ||||||
| Function / homology |  Function and homology information Function and homology informationproton-transporting two-sector ATPase complex, catalytic domain / proton motive force-driven plasma membrane ATP synthesis / proton-transporting ATP synthase activity, rotational mechanism / ATP binding / plasma membrane Similarity search - Function | ||||||
| Biological species |  Methanosarcina mazei (archaea) Methanosarcina mazei (archaea) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.7 Å | ||||||
 Authors Authors | Sundararaman, L. / Manimekalai, M.S.S. / Jeyakanthan, J. / Gruber, G. | ||||||
 Citation Citation | Journal: To be Published Title: Structure of subunit A mutants H156A, N157A and N157T of the A1AO ATP synthase Authors: Sundararaman, L. / Manimekalai, M.S.S. / Gruber, G. #1: Journal: Proteins / Year: 2009Title: Spectroscopic and crystallographic studies of the mutant R416W give insight into the nucleotide binding traits of subunit B of the A1Ao ATP synthase. Authors: Kumar, A. / Manimekalai, M.S. / Balakrishna, A.M. / Hunke, C. / Weigelt, S. / Sewald, N. / Gruber, G. #2: Journal: Acta Crystallogr.,Sect.D / Year: 2008Title: Structure of the nucleotide-binding subunit B of the energy producer A1A0 ATP synthase in complex with adenosine diphosphate. Authors: Kumar, A. / Manimekalai, M.S. / Gruber, G. #3: Journal: J.Struct.Biol. / Year: 2009Title: A second transient position of ATP on its trail to the nucleotide-binding site of subunit B of the motor protein A(1)A(0) ATP synthase. Authors: Manimekalai, M.S. / Kumar, A. / Balakrishna, A.M. / Gruber, G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3ssa.cif.gz | 216.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3ssa.ent.gz | 168.6 KB | Display | PDB format |
| PDBx/mmJSON format | 3ssa.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ss/3ssaftp://data.pdbj.org/pub/pdb/validation_reports/ss/3ssa | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3tgwC  3tivC  2c61S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 50327.242 Da / Num. of mol.: 2 / Mutation: N157T Source method: isolated from a genetically manipulated source Source: (gene. exp.) Methanosarcina mazei (archaea) / Strain: Go1 / Gene: ahaB, atpB, MM_0779 / Plasmid: pET9D-HIS6 / Production host:  References: UniProt: Q60187, H+-transporting two-sector ATPase |
|---|
-Non-polymers , 7 types, 1129 molecules 


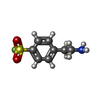



| #2: Chemical | ChemComp-GOL /  Mass: 92.094 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: C3H8O3#3: Chemical |  Mass: 35.453 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Cl#4: Chemical |  Mass: 238.278 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H22O6 / Comment: precipitant*YM Mass: 238.278 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H22O6 / Comment: precipitant*YM#5: Chemical | ChemComp-AES / |  Mass: 203.234 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H10FNO2S Mass: 203.234 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H10FNO2S#6: Chemical | ChemComp-PEG /  Mass: 106.120 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 106.120 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C4H10O3#7: Chemical | ChemComp-PGE / |  Mass: 150.173 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O4 Mass: 150.173 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O4#8: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 1106 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.27 Å3/Da / Density % sol: 45.93 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 5 Details: 30% glycerol, PEG 400, 0.1M Sodium Chloride and 0.1M Sodium citrate (pH 5.0), vapor diffusion, hanging drop, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SPring-8  / Beamline: BL12B2 / Wavelength: 1 Å / Beamline: BL12B2 / Wavelength: 1 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Feb 7, 2010 / Details: mirrors | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Redundancy: 5.4 % / Av σ(I) over netI: 20.8 / Number: 552277 / Rmerge(I) obs: 0.044 / Χ2: 0.44 / D res high: 1.7 Å / D res low: 30 Å / Num. obs: 101545 / % possible obs: 99.5 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Diffraction reflection shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.7→30 Å / Num. all: 102059 / Num. obs: 101545 / % possible obs: 99.5 % / Observed criterion σ(I): -3 / Redundancy: 5.4 % / Biso Wilson estimate: 18.934 Å2 / Rmerge(I) obs: 0.044 / Χ2: 0.44 / Net I/σ(I): 7.8 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2C61 Resolution: 1.7→13.45 Å / Cor.coef. Fo:Fc: 0.976 / Cor.coef. Fo:Fc free: 0.959 / WRfactor Rfree: 0.1916 / WRfactor Rwork: 0.1429 / Occupancy max: 1 / Occupancy min: 0.5 / FOM work R set: 0.8739 / SU B: 1.877 / SU ML: 0.062 / SU R Cruickshank DPI: 0.0898 / SU Rfree: 0.0969 / Isotropic thermal model: Isotropic / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.097 / Stereochemistry target values: Engh & Huber Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 78.41 Å2 / Biso mean: 25.3045 Å2 / Biso min: 7.48 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→13.45 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.697→1.74 Å / Total num. of bins used: 20
|
