 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3sbn | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | trichovirin I-4A in polar environment at 0.9 Angstroem | |||||||||
 Components Components | Trichovirin I-4A | |||||||||
 Keywords Keywords | ANTIBIOTIC / curved 310-helix / 3-10 HELIX / peptide antibiotic | |||||||||
| Function / homology | Trichovirin I-4A / ACETONITRILE / METHANOL / :  Function and homology information Function and homology information | |||||||||
| Biological species |  Hypocrea rufa (fungus) Hypocrea rufa (fungus) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / DIRECT METHODS / Resolution: 0.9 Å X-RAY DIFFRACTION / SYNCHROTRON / DIRECT METHODS / Resolution: 0.9 Å | |||||||||
 Authors Authors | Gessmann, R. / Axford, D. / Petratos, K. | |||||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2012 Title: Four complete turns of a curved 310-helix at atomic resolution: The crystal structure of the peptaibol trichovirin I-4A in polar environment suggests a transition to alpha-helix for membrane function Authors: Gessmann, R. / Axford, D. / Owen, R.L. / Bruckner, H. / Petratos, K. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3sbn.cif.gz | 27.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3sbn.ent.gz | 20.6 KB | Display | PDB format |
| PDBx/mmJSON format | 3sbn.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/sb/3sbnftp://data.pdbj.org/pub/pdb/validation_reports/sb/3sbn | HTTPS FTP |
|---|
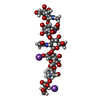
-Related structure data
| Similar structure data |
|---|







-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 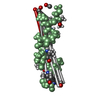
| ||||||||
| Unit cell |
| ||||||||
| Details | Author states that the biologically significant oligomerization state in apolar environment is not represented in the monomeric structure. The stoichiometry of the oligomer in the membrane is unknown. |
-Components
| #1: Protein/peptide | 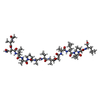  Type: Peptaibol / Class: Antibiotic / Mass: 1370.722 Da / Num. of mol.: 2 / Source method: isolated from a natural source Type: Peptaibol / Class: Antibiotic / Mass: 1370.722 Da / Num. of mol.: 2 / Source method: isolated from a natural sourceDetails: Equivalent to Harzianin HC-VI from Trichoderma harzianum Source: (natural) Hypocrea rufa (fungus) / Strain: NRRL 5243 / References: NOR: NOR00990, Trichovirin I-4A#2: Chemical |   Mass: 41.052 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H3N Mass: 41.052 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H3N#3: Chemical | 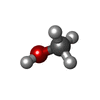  Mass: 32.042 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CH4O Mass: 32.042 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CH4O#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 16 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 16 / Source method: isolated from a natural source / Formula: H2OSequence details | AUTHOR CONFIRMS THAT THERE ARE TWO IDENTICAL ANITBOTICS UNDER DIFFERENT NAMES FROM DIFFERENT SOUCES ...AUTHOR CONFIRMS THAT THERE ARE TWO IDENTICAL ANITBOTICS | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.61 Å3/Da / Density % sol: 23.8 % |
|---|---|
| Crystal grow | Temperature: 293 K / pH: 7 Details: methanol, acetonitrile, water, pH 7.0, VAPOR DIFFUSION, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I24 / Wavelength: 0.7469 / Beamline: I24 / Wavelength: 0.7469 |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Oct 19, 2009 Details: OXFORD DANFYSIK/SESO TWO STAGE DEMAGNIFICATION USING TWO K-B PAIRS OF BIMORPH T YPE MIRRORS |
| Radiation | Monochromator: ACCEL FIXED EXIT DOUBLE CRYSTAL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.7469 Å / Relative weight: 1 |
| Reflection | Resolution: 0.9→37.3 Å / Num. obs: 13524 / % possible obs: 98.4 % / Observed criterion σ(I): 0 / Redundancy: 6.4 % / Biso Wilson estimate: 6.15 Å2 / Net I/σ(I): 13.2 |
| Reflection shell | Resolution: 0.9→0.95 Å / Redundancy: 6.2 % / Mean I/σ(I) obs: 3.1 / % possible all: 91.6 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: DIRECT METHODS / Resolution: 0.9→37.3 Å / Num. parameters: 2134 / Num. restraintsaints: 2263 / σ(F): 0 / Stereochemistry target values: ENGH & HUBER
| |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 4 / Occupancy sum hydrogen: 230 / Occupancy sum non hydrogen: 212.5 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 0.9→37.3 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
