 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3pgi | ||||||
|---|---|---|---|---|---|---|---|
| Title | Endothiapepsin in complex with a fragment | ||||||
 Components Components | Endothiapepsin | ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / HYDROLASE-HYDROLASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Cryphonectria parasitica (chestnut blight fungus) Cryphonectria parasitica (chestnut blight fungus) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Koester, H. / Heine, A. / Klebe, G. | ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2011 Title: A small nonrule of 3 compatible fragment library provides high hit rate of endothiapepsin crystal structures with various fragment chemotypes. Authors: Koster, H. / Craan, T. / Brass, S. / Herhaus, C. / Zentgraf, M. / Neumann, L. / Heine, A. / Klebe, G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3pgi.cif.gz | 77.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3pgi.ent.gz | 56.3 KB | Display | PDB format |
| PDBx/mmJSON format | 3pgi.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/pg/3pgiftp://data.pdbj.org/pub/pdb/validation_reports/pg/3pgi | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3pb5C  3pbdC  3pbzC  3pcwC  3pi0C  3pldC  3pllC  3pm4C  3pmuC  3pmyC  1oewS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |





















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 33813.855 Da / Num. of mol.: 1 / Source method: isolated from a natural source Source: (natural) Cryphonectria parasitica (chestnut blight fungus)References: UniProt: P11838, endothiapepsin |
|---|---|
| #2: Chemical | ChemComp-F41 / 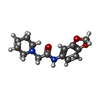  Mass: 262.304 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H18N2O3 Mass: 262.304 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H18N2O3 |
| #3: Chemical | ChemComp-GOL /   Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 239 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 239 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.43 Å3/Da / Density % sol: 49.35 % |
|---|---|
| Crystal grow | Temperature: 289 K / Method: vapor diffusion, sitting drop / pH: 4.6 Details: 0.1M NH4Ac, 0.1M Acetate Buffer, 26% PEG 4000, VAPOR DIFFUSION, SITTING DROP, temperature 277K, temperature 289K, pH 4.6 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: Cu FINE FOCUS / Wavelength: 1.54178 Å |
| Detector | Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE / Date: Jun 2, 2010 |
| Radiation | Monochromator: collimating morrors / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54178 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→30 Å / Num. all: 25348 / Num. obs: 25348 / % possible obs: 100 % / Redundancy: 3.6 % / Rsym value: 0.065 / Net I/σ(I): 26.7 |
| Reflection shell | Resolution: 1.9→1.93 Å / Redundancy: 3.2 % / Mean I/σ(I) obs: 4.3 / Num. unique all: 1213 / Rsym value: 0.287 / % possible all: 100 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1OEW Resolution: 1.9→10 Å / Num. parameters: 10515 / Num. restraintsaints: 9989 / Cross valid method: FREE R / σ(F): 0 / Stereochemistry target values: Engh & Huber Details: ANISOTROPIC SCALING APPLIED BY THE METHOD OF PARKIN, MOEZZI & HOPE, J.APPL.CRYST.28(1995)53-56
| |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 0 / Occupancy sum hydrogen: 2229 / Occupancy sum non hydrogen: 2625 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→10 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
