- PDB-3l8n: Crystal Structure of a domain of Brefeldin A-inhibited guanine nu... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 3l8n
Title
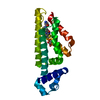
Crystal Structure of a domain of Brefeldin A-inhibited guanine nucleotide-exchange protein 2 (BrefeldinA-inhibited GEP 2) from Homo sapiens (Human). Northeast Structural Genomics Consortium target id HR5562A
Components
Brefeldin A-inhibited guanine nucleotide-exchange protein 2
Keywords
Nucleotide-binding protein / Metal-binding protein / Structural Genomics / PSI-2 / Protein Structure Initiative / Northeast Structural Genomics Consortium / NESG / Guanine-nucleotide releasing factor / Phosphoprotein
Function / homology
Function and homology information
endomembrane system organization / symmetric synapse / receptor recycling / axonemal microtubule / endosome organization / GABA receptor binding / Golgi to plasma membrane transport / regulation of ARF protein signal transduction / microtubule organizing center / myosin binding ...endomembrane system organization / symmetric synapse / receptor recycling / axonemal microtubule / endosome organization / GABA receptor binding / Golgi to plasma membrane transport / regulation of ARF protein signal transduction / microtubule organizing center / myosin binding / protein kinase A regulatory subunit binding / exocytosis / Association of TriC/CCT with target proteins during biosynthesis / asymmetric synapse / GABA-ergic synapse / guanyl-nucleotide exchange factor activity / trans-Golgi network / recycling endosome / positive regulation of tumor necrosis factor production / protein transport / presynapse / cytoplasmic vesicle / dendritic spine / intracellular signal transduction / Golgi membrane / intracellular membrane-bounded organelle / centrosome / glutamatergic synapse / perinuclear region of cytoplasm / Golgi apparatus / membrane / cytosol Similarity search - Function
Resolution: 2.8→2.91 Å / Redundancy: 1.9 % / Rmerge(I) obs: 0.343 / Rsym value: 0.29 / % possible all: 99.3
-
Processing
Software
Name
Version
Classification
ADSC
Quantum
datacollection
SHELXS
phasing
CNS
1.2
refinement
HKL-2000
datareduction
HKL-2000
datascaling
Refinement
Method to determine structure: SAD / Resolution: 2.86→17.65 Å / Rfactor Rfree error: 0.012 / Data cutoff high absF: 306158.18 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 2 / Stereochemistry target values: Engh & Huber Details: BULK SOLVENT MODEL USED. The data is twined with twinning operator= -h,-k,l and twinning fraction= 0.321.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads








 PDBj
PDBj




 Assembly
Assembly

 Sample preparation
Sample preparation / Beamline: X4A / Wavelength: 0.979 Å
/ Beamline: X4A / Wavelength: 0.979 Å Processing
Processing