THE STRUCTURE DOES NOT CONTAIN RESIDUES GLY220 TO ASP222 OF ORAE PRESENT IN THE UNP SEQUENCE. WE DO ...THE STRUCTURE DOES NOT CONTAIN RESIDUES GLY220 TO ASP222 OF ORAE PRESENT IN THE UNP SEQUENCE. WE DO NOT FIND ANY EVIDENCE FOR THESE RESIDUES FROM CLOSTRIDIUM STICKLANDII GENOMIC DNA SEQUENCING OR BY ALIGNMENT BETWEEN THE ORAE PROTEIN SEQUENCES FROM CLOSTRIDIUM STICKLANDII AND CLOSTRIDIUM DIFFICILE (ACCESSION NUMBER ZP_05349629; 79% SEQUENCE IDENTITY) WHICH ALSO SHOWS THAT THE CORRESPONDING ORAE CODING SEQUENCE FROM THE LATTER ORGANISM ALSO DOES NOT CONTAIN THE THREE AMINO ACID GID INSERT (A SIMILAR CONCLUSION CAN BE REACHED BY ALIGNMENT WITH VARIOUS THERMOANAEROBACTER SP)
-
実験情報
-
実験
実験
手法: X線回折 / 使用した結晶の数: 1
-
試料調製
結晶
マシュー密度: 2.36 Å3/Da / 溶媒含有率: 47.96 %
結晶化
温度: 294 K / 手法: 蒸気拡散法, シッティングドロップ法 / pH: 8 詳細: A protein solution (8 mg/ml 4,5-OAM, 5 mM 2-mercaptoethanol, 10 mM Tris HCl pH 8.0, 2 mM PLP, and 2 mM AdoCbl) was mixed in a 1:1 ratio with precipitant solution [(0.1 M Tris HCl, pH 8.0, 0.2 ...詳細: A protein solution (8 mg/ml 4,5-OAM, 5 mM 2-mercaptoethanol, 10 mM Tris HCl pH 8.0, 2 mM PLP, and 2 mM AdoCbl) was mixed in a 1:1 ratio with precipitant solution [(0.1 M Tris HCl, pH 8.0, 0.2 M MgCl2, 25 % (wt/vol) polyethylene glycol 2000 mono-methylether] under red light at room temperature, VAPOR DIFFUSION, SITTING DROP, temperature 294K
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Clostridium sticklandii (バクテリア)
Clostridium sticklandii (バクテリア) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード












 PDBj
PDBj 集合体
集合体






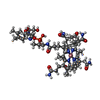
 分子量: 1330.356 Da / 分子数: 4 / 由来タイプ: 合成 / 式: C62H89CoN13O14P
分子量: 1330.356 Da / 分子数: 4 / 由来タイプ: 合成 / 式: C62H89CoN13O14P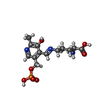
 分子量: 361.288 Da / 分子数: 4 / 由来タイプ: 合成 / 式: C13H20N3O7P
分子量: 361.288 Da / 分子数: 4 / 由来タイプ: 合成 / 式: C13H20N3O7P
 分子量: 251.242 Da / 分子数: 4 / 由来タイプ: 合成 / 式: C10H13N5O3
分子量: 251.242 Da / 分子数: 4 / 由来タイプ: 合成 / 式: C10H13N5O3 試料調製
試料調製 / ビームライン: I02 / 波長: 1.072 Å
/ ビームライン: I02 / 波長: 1.072 Å 解析
解析