 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3kgf: The structure of 3-deoxy-D-arabino-heptulosonate 7-phosphate synt... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3kgf | ||||||
|---|---|---|---|---|---|---|---|
| Title | The structure of 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase from Mycobacterium tuberculosis complexed with phenylalanine and tryptophan | ||||||
 Components Components | Probable 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase AroG | ||||||
 Keywords Keywords | TRANSFERASE / Mycobacterium tuberculosis / DAH7P synthase / Shikimate pathway / Aromatic biosynthesis / Evolutionary relationships / PHE+TRP-bound / Augmented TIM-barrel structure / Structural Genomics / Mycobacterium Tuberculosis Structural Proteomics Project / XMTB | ||||||
| Function / homology |  Function and homology information Function and homology information3-deoxy-7-phosphoheptulonate synthase / 3-deoxy-7-phosphoheptulonate synthase activity / Chorismate via Shikimate Pathway / chorismate biosynthetic process / aromatic amino acid biosynthetic process / amino acid biosynthetic process / peptidoglycan-based cell wall / protein homooligomerization / manganese ion binding / plasma membrane / cytosol Similarity search - Function | ||||||
| Biological species |   Mycobacterium tuberculosis (bacteria) Mycobacterium tuberculosis (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Parker, E.J. / Jameson, G.B. / Jiao, W. / Webby, C.J. / Baker, E.N. / Baker, H.M. / Mycobacterium Tuberculosis Structural Proteomics Project (XMTB) | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2010 Title: Synergistic allostery, a sophisticated regulatory network for the control of aromatic amino acid biosynthesis in Mycobacterium tuberculosis Authors: Webby, C.J. / Jiao, W. / Hutton, R.D. / Blackmore, N.J. / Baker, H.M. / Baker, E.N. / Jameson, G.B. / Parker, E.J. #1: Journal: J.Mol.Biol. / Year: 2005Title: The structure of 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase from Mycobacterium tuberculosis reveals a common catalytic scaffold and ancestry for type I and type II enzymes Authors: Webby, C.J. / Baker, H.M. / Lott, J.S. / Baker, E.N. / Parker, E.J. #2: Journal: Biochemistry / Year: 2005Title: Substrate ambiguity and crystal structure of Pyrococcus furiosus 3-deoxy-D-arabino-heptulosonate-7-phosphate synthase: an ancestral 3-deoxyald-2-ulosonate-phosphate synthase? Authors: Schofield, L.R. / Anderson, B.F. / Patchett, M.L. / Norris, G.E. / Jameson, G.B. / Parker, E.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3kgf.cif.gz | 344 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3kgf.ent.gz | 272.3 KB | Display | PDB format |
| PDBx/mmJSON format | 3kgf.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/kg/3kgfftp://data.pdbj.org/pub/pdb/validation_reports/kg/3kgf | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3nudC  3nueC  3nv8C  2b7oS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data | |
| Other databases |










-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: 1 / Ens-ID: 1 / End auth comp-ID: ASP / End label comp-ID: ASP / Refine code: 5
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 50828.395 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Mycobacterium tuberculosis (bacteria) / Strain: H37Rv / Gene: Rv2178c / Plasmid: PproExHTa / Production host: References: UniProt: O53512, 3-deoxy-7-phosphoheptulonate synthase |
|---|
-Non-polymers , 8 types, 801 molecules 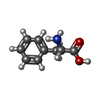
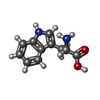






| #2: Chemical |  Type: L-peptide linking / Mass: 165.189 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C9H11NO2 Type: L-peptide linking / Mass: 165.189 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C9H11NO2#3: Chemical |  Type: L-peptide linking / Mass: 204.225 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C11H12N2O2 Type: L-peptide linking / Mass: 204.225 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C11H12N2O2#4: Chemical | ChemComp-GOL /  Mass: 92.094 Da / Num. of mol.: 16 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 16 / Source method: obtained synthetically / Formula: C3H8O3#5: Chemical |  Mass: 94.971 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: PO4#6: Chemical |  Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn#7: Chemical |  Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl#8: Chemical |  Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#9: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 771 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.95 Å3/Da / Density % sol: 68.84 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: 20mM BTP, 150mM NaCl, 0.5mM TCEP, 0.005%(v/v) Thesit, 0.2mM PEP, 0.1mM MnCl2, 0.1M Na HEPES, 0.8M NaK phosphate, pH 7.5, VAPOR DIFFUSION, SITTING DROP, temperature 291.0K |
-Data collection
| Diffraction | Mean temperature: 120 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 / Wavelength: 1.542 Å |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Dec 14, 2005 / Details: Osmic Blue Mirrors |
| Radiation | Monochromator: OSMIC BLUE MIRRORS / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.542 Å / Relative weight: 1 |
| Reflection | Resolution: 2→39.5 Å / Num. all: 105425 / Num. obs: 105425 / % possible obs: 98.2 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 2.07 % / Biso Wilson estimate: 37.5 Å2 / Rmerge(I) obs: 0.064 / Net I/σ(I): 8.2 |
| Reflection shell | Resolution: 2→2.07 Å / Redundancy: 2.38 % / Rmerge(I) obs: 0.381 / Mean I/σ(I) obs: 2.1 / Num. unique all: 10487 / % possible all: 87.2 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2b7o Resolution: 2→32.67 Å / Cor.coef. Fo:Fc: 0.964 / Cor.coef. Fo:Fc free: 0.953 / Occupancy max: 1 / Occupancy min: 0.2 / SU B: 5.535 / SU ML: 0.082 / Isotropic thermal model: isotropic / Cross valid method: THROUGHOUT / ESU R: 0.118 / ESU R Free: 0.113 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. MN is chain A is present in 75% occupancy, while in chain B its occupancy is 50%. The water that is observed coordinated to the MN ion in ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. MN is chain A is present in 75% occupancy, while in chain B its occupancy is 50%. The water that is observed coordinated to the MN ion in chain A is displaced to more than 4 Angstroms away from the MN in chain B. Atom OE1 of GLU411 is weakly coordinated at distances greater than 2.6 Angstroms for both MN sites. Because the occupancy of MN in chain B is only two-thirds that of the MN in chain A, MN-ligand distances in chain B are ~0.2 A longer than the corresponding distances in chain A.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 84.1 Å2 / Biso mean: 25.418 Å2 / Biso min: 9.29 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→32.67 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Dom-ID: 1 / Auth asym-ID: A / Ens-ID: 1 / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.052 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
