 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3kaf: Structure-guided design of alpha-amino acid-derived Pin1 inhibitors -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3kaf | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure-guided design of alpha-amino acid-derived Pin1 inhibitors | ||||||
 Components Components | Peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 | ||||||
 Keywords Keywords | ISOMERASE / SBDD / PPIASE / ROTAMASE / SMALL MOLECULE / Proline directed kinase / cell cycle / Oncogenic transformation / Nucleus / Phosphoprotein | ||||||
| Function / homology |  Function and homology information Function and homology informationcis-trans isomerase activity / phosphothreonine residue binding / negative regulation of cell motility / negative regulation of brown fat cell differentiation / regulation of protein localization to nucleus / mitogen-activated protein kinase kinase binding / ubiquitin ligase activator activity / GTPase activating protein binding / : / protein peptidyl-prolyl isomerization ...cis-trans isomerase activity / phosphothreonine residue binding / negative regulation of cell motility / negative regulation of brown fat cell differentiation / regulation of protein localization to nucleus / mitogen-activated protein kinase kinase binding / ubiquitin ligase activator activity / GTPase activating protein binding / : / protein peptidyl-prolyl isomerization / regulation of mitotic nuclear division / negative regulation of SMAD protein signal transduction / PI5P Regulates TP53 Acetylation / negative regulation of amyloid-beta formation / cytoskeletal motor activity / phosphoserine residue binding / RHO GTPases Activate NADPH Oxidases / postsynaptic cytosol / Rho protein signal transduction / regulation of cytokinesis / peptidylprolyl isomerase / peptidyl-prolyl cis-trans isomerase activity / Negative regulators of DDX58/IFIH1 signaling / negative regulation of transforming growth factor beta receptor signaling pathway / regulation of protein stability / phosphoprotein binding / negative regulation of protein catabolic process / negative regulation of ERK1 and ERK2 cascade / positive regulation of protein phosphorylation / synapse organization / beta-catenin binding / protein destabilization / tau protein binding / ISG15 antiviral mechanism / neuron differentiation / positive regulation of canonical Wnt signaling pathway / regulation of gene expression / midbody / cellular response to hypoxia / Regulation of TP53 Activity through Phosphorylation / response to hypoxia / nuclear speck / protein stabilization / ciliary basal body / glutamatergic synapse / positive regulation of transcription by RNA polymerase II / nucleoplasm / nucleus / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MIR / Resolution: 2.3 Å X-RAY DIFFRACTION / MIR / Resolution: 2.3 Å | ||||||
 Authors Authors | Baker, L.M. / Dokurno, P. / Robinson, D.A. / Surgenor, A.E. / Murray, J.B. / Potter, A.J. / Moore, J.D. | ||||||
 Citation Citation | Journal: Bioorg.Med.Chem.Lett. / Year: 2010 Title: Structure-guided design of alpha-amino acid-derived Pin1 inhibitors Authors: Potter, A.J. / Ray, S. / Gueritz, L. / Nunns, C.L. / Bryant, C.J. / Scrace, S.F. / Matassova, N. / Baker, L.M. / Dokurno, P. / Robinson, D.A. / Surgenor, A.E. / Davis, B. / Murray, J.B. / ...Authors: Potter, A.J. / Ray, S. / Gueritz, L. / Nunns, C.L. / Bryant, C.J. / Scrace, S.F. / Matassova, N. / Baker, L.M. / Dokurno, P. / Robinson, D.A. / Surgenor, A.E. / Davis, B. / Murray, J.B. / Richardson, C.M. / Moore, J.D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3kaf.cif.gz | 45.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3kaf.ent.gz | 31.1 KB | Display | PDB format |
| PDBx/mmJSON format | 3kaf.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ka/3kafftp://data.pdbj.org/pub/pdb/validation_reports/ka/3kaf | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3kabC  3kacC  3kadC  3kagC  3kahC  3kaiC  3kceC  1pinS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 18467.475 Da / Num. of mol.: 1 / Mutation: R14A, Q131A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: PIN1 / Plasmid: PET28A / Production host:  |
|---|---|
| #2: Chemical | ChemComp-4D9 / 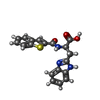  Mass: 365.406 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H15N3O3S Mass: 365.406 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H15N3O3S |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 48 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 48 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.86 Å3/Da / Density % sol: 56.97 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: 2.2M Ammonium sulphate, 0.1M HEPES buffer, 1% PEG 400, 5mM DTT, pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH3R / Wavelength: 1.5418 Å |
| Detector | Type: RIGAKU RAXIS IV++ / Detector: IMAGE PLATE / Date: Nov 3, 2006 / Details: mirrors |
| Radiation | Monochromator: mirrors / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→27.6 Å / Num. all: 9395 / Num. obs: 9395 / % possible obs: 96.2 % / Observed criterion σ(F): 1 / Observed criterion σ(I): 1 / Redundancy: 2.9 % / Rmerge(I) obs: 0.081 / Net I/σ(I): 4.6 |
| Reflection shell | Resolution: 2.3→2.38 Å / Redundancy: 2.8 % / Rmerge(I) obs: 0.438 / Mean I/σ(I) obs: 1.3 / Num. unique all: 885 / % possible all: 99.8 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MIR Starting model: PDB ENTRY 1PIN Resolution: 2.3→27.6 Å / Cor.coef. Fo:Fc: 0.945 / Cor.coef. Fo:Fc free: 0.898 / SU B: 9.04 / SU ML: 0.213 / Cross valid method: THROUGHOUT / ESU R: 0.296 / ESU R Free: 0.274 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 46.677 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→27.6 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.3→2.36 Å / Total num. of bins used: 20
|
