 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3jzi: Crystal structure of biotin carboxylase from E. Coli in complex w... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3jzi | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of biotin carboxylase from E. Coli in complex with benzimidazole series | ||||||
 Components Components | Biotin carboxylase | ||||||
 Keywords Keywords | LIGASE / Biotin carboxylase / AccC / Acetyl coenzyme-A carboxylase / ACCase / ATP-binding / Biotin / Fatty acid biosynthesis / Lipid synthesis / Nucleotide-binding | ||||||
| Function / homology |  Function and homology information Function and homology informationbiotin carboxylase / malonyl-CoA biosynthetic process / biotin carboxylase activity / acetyl-CoA carboxylase activity / negative regulation of fatty acid biosynthetic process / fatty acid biosynthetic process / protein homodimerization activity / ATP binding / metal ion binding / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.31 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.31 Å | ||||||
 Authors Authors | Cheng, C. / Shipps, G.W. / Yang, Z. / Sun, B. / Kawahata, N. / Soucy, K. / Soriano, A. / Orth, P. / Xiao, L. / Mann, P. / Black, T. | ||||||
 Citation Citation | Journal: Bioorg.Med.Chem.Lett. / Year: 2009 Title: Discovery and optimization of antibacterial AccC inhibitors. Authors: Cheng, C.C. / Shipps, G.W. / Yang, Z. / Sun, B. / Kawahata, N. / Soucy, K.A. / Soriano, A. / Orth, P. / Xiao, L. / Mann, P. / Black, T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3jzi.cif.gz | 192.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3jzi.ent.gz | 149.9 KB | Display | PDB format |
| PDBx/mmJSON format | 3jzi.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/jz/3jziftp://data.pdbj.org/pub/pdb/validation_reports/jz/3jzi | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3jzfC  1bncS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 53507.059 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: P24182, biotin carboxylase, acetyl-CoA carboxylase #2: Chemical | 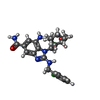  Mass: 441.954 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C23H28ClN5O2 Mass: 441.954 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C23H28ClN5O2#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 400 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 400 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.6 Å3/Da / Density % sol: 52.75 % Description: Use of the IMCA-CAT beamline 17-ID at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with the ...Description: Use of the IMCA-CAT beamline 17-ID at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with the Center for Advanced Radiation Sources at the University of Chicago. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, sitting drop / pH: 8 Details: Co-crystallization. 8-10% PEG6000, 100mM Tris-HCl., VAPOR DIFFUSION, SITTING DROP, temperature 295K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 17-ID / Wavelength: 1 Å / Beamline: 17-ID / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 210 / Detector: CCD / Date: Jan 1, 2006 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→20 Å / Num. obs: 47651 / % possible obs: 97.5 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Biso Wilson estimate: 43.2 Å2 |
| Reflection shell | Resolution: 2.3→2.38 Å / % possible all: 98.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1BNC Resolution: 2.31→19.97 Å / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 39.13 Å2
| ||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.27 Å | ||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.31→19.97 Å
| ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.31→2.37 Å / Total num. of bins used: 20
|
