 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
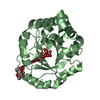
Yorodumi- PDB-3hd0: Crystal structure of Tm1865, an Endonuclease V from Thermotoga Ma... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3hd0 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Tm1865, an Endonuclease V from Thermotoga Maritima | ||||||
 Components Components | Endonuclease V | ||||||
 Keywords Keywords | HYDROLASE / structural genomics / ISFI / RNaseH SUPERFAMILY / Endonuclease V / PSI-2 / Protein Structure Initiative / Integrated Center for Structure and Function Innovation / Cytoplasm / DNA damage / DNA repair / Endonuclease / Magnesium / Nuclease | ||||||
| Function / homology |  Function and homology information Function and homology informationdeoxyribonuclease V / deoxyribonuclease V activity / RNA endonuclease activity producing 5'-phosphomonoesters, hydrolytic mechanism / single-stranded RNA binding / DNA repair / magnesium ion binding / cytoplasm Similarity search - Function | ||||||
| Biological species |   Thermotoga maritima (bacteria) Thermotoga maritima (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.7 Å | ||||||
 Authors Authors | Utepbergenov, D. / Cooper, D.R. / Derewenda, U. / Derewenda, Z.S. / Integrated Center for Structure and Function Innovation (ISFI) | ||||||
 Citation Citation | Journal: To be Published Title: Crystal structure of Tm1865, an Endonuclease V from Thermotoga Maritima Authors: Utepbergenov, D. / Cooper, D.R. / Derewenda, U. / Derewenda, Z.S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3hd0.cif.gz | 271.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3hd0.ent.gz | 222 KB | Display | PDB format |
| PDBx/mmJSON format | 3hd0.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hd/3hd0ftp://data.pdbj.org/pub/pdb/validation_reports/hd/3hd0 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data | |
|---|---|
| Other databases |








-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological unit is a monomer. |
-Components
| #1: Protein | Mass: 27332.053 Da / Num. of mol.: 3 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Thermotoga maritima (bacteria) / Gene: nfi, TM_1865 / Production host: #2: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 221 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 221 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.84 Å3/Da / Density % sol: 33 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 8.5 Details: Crystallization drop was 1:1 mixture of protein dissolved in 20 mM Tris pH 8.5, 100 mM NaCl and 24 w/v% PEG 1500, 20% glycerol. The crystallization reservoir was 1.5 M NaCl, VAPOR DIFFUSION, ...Details: Crystallization drop was 1:1 mixture of protein dissolved in 20 mM Tris pH 8.5, 100 mM NaCl and 24 w/v% PEG 1500, 20% glycerol. The crystallization reservoir was 1.5 M NaCl, VAPOR DIFFUSION, SITTING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 22-ID / Wavelength: 0.97876, 0.97860, 0.97243 / Beamline: 22-ID / Wavelength: 0.97876, 0.97860, 0.97243 | ||||||||||||
| Detector | Type: MARMOSAIC 300 mm CCD / Detector: CCD / Date: Jun 27, 2007 | ||||||||||||
| Radiation | Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||
| Radiation wavelength |
| ||||||||||||
| Reflection | Resolution: 2.7→50 Å / Num. obs: 17147 / % possible obs: 99.8 % / Redundancy: 5.3 % / Rmerge(I) obs: 0.131 / Net I/σ(I): 15.333 | ||||||||||||
| Reflection shell | Resolution: 2.7→2.8 Å / Redundancy: 4.9 % / Rmerge(I) obs: 0.453 / Mean I/σ(I) obs: 3.27 / % possible all: 99.8 |
-Phasing
| Phasing | Method: MAD | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phasing set |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing MAD set |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing MAD set site |
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.7→50 Å / Cor.coef. Fo:Fc: 0.951 / Cor.coef. Fo:Fc free: 0.882 / Occupancy max: 1 / Occupancy min: 1 / SU B: 26.597 / SU ML: 0.274 / Cross valid method: THROUGHOUT / ESU R Free: 0.41 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 28.281 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.7→50 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.703→2.773 Å / Total num. of bins used: 20
|
