 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3fiv: CRYSTAL STRUCTURE OF FELINE IMMUNODEFICIENCY VIRUS PROTEASE COMPL... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3fiv | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF FELINE IMMUNODEFICIENCY VIRUS PROTEASE COMPLEXED WITH A SUBSTRATE | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / ASPARTIC PROTEASE / RETROVIRAL PROTEASE / HYDROLASE-HYDROLASE INHIBITOR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationdUTP catabolic process / dUMP biosynthetic process / dUTP diphosphatase / dUTP diphosphatase activity / Hydrolases; Acting on peptide bonds (peptidases); Aspartic endopeptidases / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / DNA integration / viral genome integration into host DNA ...dUTP catabolic process / dUMP biosynthetic process / dUTP diphosphatase / dUTP diphosphatase activity / Hydrolases; Acting on peptide bonds (peptidases); Aspartic endopeptidases / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / DNA integration / viral genome integration into host DNA / RNA-directed DNA polymerase / establishment of integrated proviral latency / RNA stem-loop binding / RNA-directed DNA polymerase activity / RNA-DNA hybrid ribonuclease activity / Transferases; Transferring phosphorus-containing groups; Nucleotidyltransferases / DNA recombination / aspartic-type endopeptidase activity / Hydrolases; Acting on ester bonds / symbiont entry into host cell / magnesium ion binding / proteolysis / DNA binding / zinc ion binding Similarity search - Function | ||||||
| Biological species |  Feline immunodeficiency virus Feline immunodeficiency virus | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.85 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.85 Å | ||||||
 Authors Authors | Schalk-Hihi, C. / Lubkowski, J. / Zdanov, A. / Wlodawer, A. / Gustchina, A. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1997 Title: Crystal structures of the inactive D30N mutant of feline immunodeficiency virus protease complexed with a substrate and an inhibitor. Authors: Laco, G.S. / Schalk-Hihi, C. / Lubkowski, J. / Morris, G. / Zdanov, A. / Olson, A. / Elder, J.H. / Wlodawer, A. / Gustchina, A. #1: Journal: Nat.Struct.Biol. / Year: 1995Title: Structure of an Inhibitor Complex of the Proteinase from Feline Immunodeficiency Virus Authors: Wlodawer, A. / Gustchina, A. / Reshetnikova, L. / Lubkowski, J. / Zdanov, A. / Hui, K.Y. / Angleton, E.L. / Farmerie, W.G. / Goodenow, M.M. / Bhatt, D. / Zhang, L. / Dunn, B.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3fiv.cif.gz | 63.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3fiv.ent.gz | 49.2 KB | Display | PDB format |
| PDBx/mmJSON format | 3fiv.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 3fiv_validation.pdf.gz | 447.7 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 3fiv_full_validation.pdf.gz | 455.9 KB | Display | |
| Data in XML | 3fiv_validation.xml.gz | 14.2 KB | Display | |
| Data in CIF | 3fiv_validation.cif.gz | 18.9 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fi/3fivftp://data.pdbj.org/pub/pdb/validation_reports/fi/3fiv | HTTPS FTP |
-Related structure data
| Related structure data |  2fivC 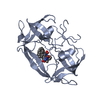 1fivS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 13280.297 Da / Num. of mol.: 2 / Mutation: D30N Source method: isolated from a genetically manipulated source Source: (gene. exp.) Feline immunodeficiency virus / Genus: Lentivirus / Cell line: BL21 / Plasmid: BL21 / Production host:  #2: Protein/peptide | 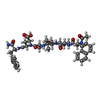  Type: Peptide-like / Class: Inhibitor / Mass: 849.007 Da / Num. of mol.: 2 / Source method: obtained synthetically Type: Peptide-like / Class: Inhibitor / Mass: 849.007 Da / Num. of mol.: 2 / Source method: obtained syntheticallyReferences: N-acetyl-3-(naphthalen-1-yl)-L-alanyl-L-valyl-L-leucyl-L-alanyl-L-alpha-glutamyl-3-naphthalen-1-yl- L-alaninamide #3: Chemical |   Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 148 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 148 / Source method: isolated from a natural source / Formula: H2OCompound details | THE INHIBITOR ACE-ALN-VAL-LEU-ALA-GLU-ALN-NH2 IS DISORDERED. IT APPEARS IN TWO ORIENTATIONS (CHAINS ...THE INHIBITOR ACE-ALN-VAL-LEU-ALA-GLU-ALN-NH2 IS DISORDERED | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.05 Å3/Da / Density % sol: 37.61 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 5.6 Details: PROTEIN WAS CRYSTALLIZED FROM 2.0 M AMMONIUM SULFATE, 0.1 M SODIUM ACETATE, PH=5.6. PROTEIN CONCENTRATION, 5.0 MG/ML. CRYSTALLIZATION METHOD: HANGING DROP VAPOR DIFFUSION | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 7 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: ENRAF-NONIUS FR591 / Wavelength: 1.5418 |
| Detector | Type: MACSCIENCE / Detector: IMAGE PLATE / Date: Apr 23, 1996 / Details: MIRRORS |
| Radiation | Monochromator: NI FILTER / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→20 Å / Num. obs: 40465 / % possible obs: 87.2 % / Observed criterion σ(I): 1 / Redundancy: 2.32 % / Rsym value: 0.091 / Net I/σ(I): 16 |
| Reflection shell | Resolution: 1.8→1.83 Å / Redundancy: 1.65 % / Mean I/σ(I) obs: 2.59 / Rsym value: 0.42 / % possible all: 60.8 |
| Reflection | *PLUS Num. obs: 17411 / Num. measured all: 40465 / Rmerge(I) obs: 0.091 |
| Reflection shell | *PLUS % possible obs: 68.5 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1FIV Resolution: 1.85→8 Å / Data cutoff high absF: 10000000 / Data cutoff low absF: 0.001 / Isotropic thermal model: RESTRAINED / σ(F): 3 Details: HUBER AND ENGH PARAMETERS WERE USED IN THE REFINEMENT. FOR NON-STANDARD RESIDUES (CYO, ALN, NAM, ACE, SO4) LIBRARIES BASED ON EXISTING PARAMETERS WERE CREATED BY SIMILARITY. THE X-RAY DATA ...Details: HUBER AND ENGH PARAMETERS WERE USED IN THE REFINEMENT. FOR NON-STANDARD RESIDUES (CYO, ALN, NAM, ACE, SO4) LIBRARIES BASED ON EXISTING PARAMETERS WERE CREATED BY SIMILARITY. THE X-RAY DATA ARE COMPATIBLE WITH THE SPACE GROUP P31 2 1. THE LOWER SYMMETRY SPACE GROUP WAS UTILIZED IN REFINEMENT FOR TECHNICAL REASONS. THE CHAINS A AND B (AND I AND J) ARE THUS RELATED BY NON-CRYSTALLOGRAPHIC SYMMETRY, EQUIVALENT TO TWO-FOLD AXIS DIFFERENTIATING THE TWO SPACE GROUPS. THE Z VALUE IS GIVEN ON THE ASSUMPTION OF IDENTITY OF CHAINS A AND B.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 28 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.85→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: RESTRAINTS / Rms dev Biso : 1.35 Å2 / Rms dev position: 0.005 Å / Weight Biso : 0.5 / Weight position: 1000 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.85→1.93 Å / Total num. of bins used: 8 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
