 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3cxm: Leishmania naiffi uracil-DNA glycosylase in complex with 5-bromouracil -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3cxm | ||||||
|---|---|---|---|---|---|---|---|
| Title | Leishmania naiffi uracil-DNA glycosylase in complex with 5-bromouracil | ||||||
 Components Components | Uracil-DNA glycosylase | ||||||
 Keywords Keywords | HYDROLASE / base excision repair / BER / DNA damage repair / Leishmania / MSGPP / SGPP / glycosylase / 5-bromouracil / Structural Genomics / Structural Genomics of Pathogenic Protozoa Consortium / DNA repair / Glycosidase / PSI-2 / Protein Structure Initiative | ||||||
| Function / homology |  Function and homology information Function and homology informationbase-excision repair, AP site formation via deaminated base removal / uracil-DNA glycosylase / uracil DNA N-glycosylase activity / mitochondrion / nucleus Similarity search - Function | ||||||
| Biological species |  Leishmania naiffi (eukaryote) Leishmania naiffi (eukaryote) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.5 Å | ||||||
 Authors Authors | Larson, E.T. / Merritt, E.A. / Structural Genomics of Pathogenic Protozoa Consortium (SGPP) | ||||||
 Citation Citation | Journal: To be Published Title: Structures of Leishmania naiffi uracil-DNA glycosylase in complex with several ligands identified with fragment cocktail crystallography. Authors: Larson, E.T. / Merritt, E.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3cxm.cif.gz | 73.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3cxm.ent.gz | 53 KB | Display | PDB format |
| PDBx/mmJSON format | 3cxm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cx/3cxmftp://data.pdbj.org/pub/pdb/validation_reports/cx/3cxm | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1euiS S: Starting model for refinement |
|---|---|
| Similar structure data | |
| Other databases |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 30285.660 Da / Num. of mol.: 1 / Fragment: Residues 129-373 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Leishmania naiffi (eukaryote) / Gene: similar to LbrM18_V2.0540 / Plasmid: AVA0421 / Species (production host): Escherichia coli / Production host:  References: UniProt: D0VWU0*PLUS, Hydrolases; Glycosylases; Hydrolysing N-glycosyl compounds |
|---|
-Non-polymers , 5 types, 244 molecules 



| #2: Chemical | ChemComp-BR /  Mass: 79.904 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Br Mass: 79.904 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Br |
|---|---|
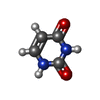
| #3: Chemical | ChemComp-URB /  Mass: 190.983 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H3BrN2O2 Mass: 190.983 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H3BrN2O2 |
| #4: Chemical | ChemComp-URA /  Mass: 112.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H4N2O2 Mass: 112.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H4N2O2 |
| #5: Chemical | ChemComp-GOL /  Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 |
| #6: Water | ChemComp-HOH / Mass: 18.015 Da / Num. of mol.: 240 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Sequence details | 1. THE SEQUENCE OF THIS PROTEIN WAS NOT AVAILABLE AT THE UNIPROT KNOWLEDGEBASE DATABASE (UNIPROTKB) ...1. THE SEQUENCE OF THIS PROTEIN WAS NOT AVAILABLE AT THE UNIPROT KNOWLEDGEB |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.86 Å3/Da / Density % sol: 56.96 % Description: 1. The Blu-ice software package was used for data collection. The reference is: McPhillips T.M., McPhillips S.E., Chiu H.J., Cohen A.E., Deacon A.M., Ellis P.J., Garman E., Gonzalez A., ...Description: 1. The Blu-ice software package was used for data collection. The reference is: McPhillips T.M., McPhillips S.E., Chiu H.J., Cohen A.E., Deacon A.M., Ellis P.J., Garman E., Gonzalez A., Sauter N.K., Phizackerley R.P., Soltis S.M., Kuhn P., Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. J.Synchrotron Radiat. 2002 Nov 1, 9(Pt 6):401-6. Epub 2002 Nov 1. PMID: 12409628. 2. The TLS motion determination server (http://skuld.bmsc.washington.edu/~tlsmd/) was used for selection of the TLS groups used in refinement. The reference is: Painter J. and Merritt E.A., TLSMD web server for the generation of multi-group TLS models. J.Appl.Cryst. 2006 Feb, 39(Pt 1):109-111. Epub 2006 Jan 12. |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 8.5 Details: 1.3 M Potassium phosphate dibasic, 0.1 M Sodium acetate pH 4.5, 5 mM DTT, pH 8.5, VAPOR DIFFUSION, SITTING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL9-2 / Wavelength: 0.91724 Å / Beamline: BL9-2 / Wavelength: 0.91724 Å |
| Detector | Type: MARMOSAIC 325 mm CCD / Detector: CCD / Date: Mar 28, 2007 / Details: Mirrors |
| Radiation | Monochromator: Double crystal Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.91724 Å / Relative weight: 1 |
| Reflection | Resolution: 1.5→50 Å / Num. obs: 52821 / % possible obs: 97.2 % / Redundancy: 10.2 % / Biso Wilson estimate: 21.8 Å2 / Rmerge(I) obs: 0.045 / Χ2: 0.978 / Net I/σ(I): 14.3 |
| Reflection shell | Resolution: 1.5→1.55 Å / Redundancy: 5.3 % / Rmerge(I) obs: 0.62 / Mean I/σ(I) obs: 1.5 / Num. unique all: 4325 / Χ2: 0.979 / % possible all: 79.9 |
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR |
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 1EUI Resolution: 1.5→37.66 Å / Cor.coef. Fo:Fc: 0.982 / Cor.coef. Fo:Fc free: 0.977 / WRfactor Rfree: 0.155 / WRfactor Rwork: 0.133 / SU B: 2.276 / SU ML: 0.042 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.05 / ESU R Free: 0.053 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: 1. ATOM RECORD CONTAINS RESIDUAL B FACTORS ONLY. 2. HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 15.353 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.5→37.66 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
