Mass: 18.015 Da / Num. of mol.: 84 / Source method: isolated from a natural source / Formula: H2O
Has ligand of interest
Y
Has protein modification
Y
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 1.96 Å3/Da / Density % sol: 37.25 %
Crystal grow
Temperature: 295 K / Method: batch mode / pH: 4.7 Details: HEWL CO-CRYSTALLIZED WITH 150 MM EXCESS N-ACETYLGLUCOSAMINE IN 1 ML 0.05 M SODIUM ACETATE AND 1 ML 10% SODIUM CHLORIDE, PH 4.7, BATCH, TEMPERATURE 295K Temp details: Room temperature
-
Data collection
Diffraction
Mean temperature: 100 K / Serial crystal experiment: N
Diffraction source
Source: ROTATING ANODE / Type: Cu FINE FOCUS / Wavelength: 1.54184 Å
Detector
Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Feb 1, 2011 / Details: OSMIC CONFOCAL MAX-FLUX, BLUE
Radiation
Monochromator: confocal mirror optics / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 1.54184 Å / Relative weight: 1
Reflection
Resolution: 2→19.606 Å / Num. obs: 7468 / % possible obs: 91.5 % / Redundancy: 12.46 % / Biso Wilson estimate: 23.8 Å2 / CC1/2: 0.996 / CC star: 0.999 / Rpim(I) all: 0.073 / Rrim(I) all: 0.253 / Rsym value: 0.243 / Χ2: 1 / Net I/av σ(I): 11.17 / Net I/σ(I): 8.15
Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2→19.606 Å / Cor.coef. Fo:Fc: 0.956 / Cor.coef. Fo:Fc free: 0.928 / SU B: 13.461 / SU ML: 0.186 / Cross valid method: THROUGHOUT / ESU R: 0.306 / ESU R Free: 0.226 Details: Hydrogens have been added in their riding positions
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.2595
753
10.125 %
RANDOM
Rwork
0.2038
6684
-
-
all
0.209
-
-
-
obs
-
7437
91.431 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK BULK SOLVENT
Displacement parameters
Biso mean: 21.853 Å2
Baniso -1
Baniso -2
Baniso -3
1-
1.186 Å2
-0 Å2
-0 Å2
2-
-
1.186 Å2
-0 Å2
3-
-
-
-2.371 Å2
Refinement step
Cycle: LAST / Resolution: 2→19.606 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
1001
0
24
84
1109
Refine LS restraints
Refine-ID
Type
Dev ideal
Dev ideal target
Number
X-RAY DIFFRACTION
r_bond_refined_d
0.005
0.016
1040
X-RAY DIFFRACTION
r_bond_other_d
0.001
0.016
946
X-RAY DIFFRACTION
r_angle_refined_deg
0.964
1.788
1428
X-RAY DIFFRACTION
r_angle_other_deg
0.349
1.576
2158
X-RAY DIFFRACTION
r_dihedral_angle_1_deg
6.594
5.396
139
X-RAY DIFFRACTION
r_dihedral_angle_3_deg
12.648
10
166
X-RAY DIFFRACTION
r_dihedral_angle_6_deg
14.998
10
50
X-RAY DIFFRACTION
r_chiral_restr
0.038
0.2
149
X-RAY DIFFRACTION
r_gen_planes_refined
0.003
0.02
1279
X-RAY DIFFRACTION
r_gen_planes_other
0.001
0.02
281
X-RAY DIFFRACTION
r_nbd_refined
0.197
0.2
264
X-RAY DIFFRACTION
r_symmetry_nbd_other
0.187
0.2
904
X-RAY DIFFRACTION
r_nbtor_refined
0.167
0.2
530
X-RAY DIFFRACTION
r_symmetry_nbtor_other
0.078
0.2
540
X-RAY DIFFRACTION
r_xyhbond_nbd_refined
0.13
0.2
65
X-RAY DIFFRACTION
r_metal_ion_refined
0.017
0.2
1
X-RAY DIFFRACTION
r_symmetry_nbd_refined
0.101
0.2
9
X-RAY DIFFRACTION
r_nbd_other
0.135
0.2
53
X-RAY DIFFRACTION
r_symmetry_xyhbond_nbd_refined
0.099
0.2
12
X-RAY DIFFRACTION
r_mcbond_it
1.02
1.957
515
X-RAY DIFFRACTION
r_mcbond_other
1.02
1.957
515
X-RAY DIFFRACTION
r_mcangle_it
1.757
3.565
652
X-RAY DIFFRACTION
r_mcangle_other
1.754
3.563
652
X-RAY DIFFRACTION
r_scbond_it
1.644
2.205
525
X-RAY DIFFRACTION
r_scbond_other
1.643
2.205
526
X-RAY DIFFRACTION
r_scangle_it
2.837
3.98
779
X-RAY DIFFRACTION
r_scangle_other
2.835
3.979
780
X-RAY DIFFRACTION
r_lrange_it
5.28
21.152
1307
X-RAY DIFFRACTION
r_lrange_other
5.265
20.547
1293
LS refinement shell
Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Rfactor all
Num. reflection all
Fsc free
Fsc work
% reflection obs (%)
WRfactor Rwork
2-2.052
0.293
45
0.319
544
0.317
589
0.925
0.921
100
0.297
2.052-2.107
0.32
61
0.284
507
0.288
568
0.921
0.941
100
0.257
2.107-2.168
0.355
53
0.277
490
0.285
543
0.897
0.946
100
0.247
2.168-2.234
0.352
57
0.276
472
0.284
529
0.925
0.947
100
0.24
2.234-2.306
0.279
53
0.253
478
0.256
531
0.947
0.955
100
0.212
2.306-2.386
0.369
46
0.262
449
0.271
495
0.907
0.953
100
0.214
2.386-2.474
0.318
66
0.253
426
0.263
492
0.94
0.954
100
0.206
2.474-2.573
0.295
43
0.22
417
0.227
460
0.94
0.967
100
0.173
2.573-2.686
0.372
54
0.226
405
0.243
459
0.928
0.966
100
0.191
2.686-2.814
0.402
23
0.222
207
0.239
447
0.935
0.966
51.4541
0.175
2.814-2.963
0.235
23
0.212
224
0.215
403
0.966
0.972
61.2903
0.177
2.963-3.138
0.249
34
0.201
368
0.205
402
0.961
0.972
100
0.169
3.138-3.349
0.34
8
0.176
66
0.194
385
0.941
0.98
19.2208
0.15
3.349-3.609
0.234
44
0.181
302
0.188
346
0.958
0.979
100
0.173
3.609-3.94
0.219
32
0.155
294
0.16
326
0.963
0.984
100
0.15
3.94-4.383
0.129
29
0.14
271
0.138
300
0.986
0.987
100
0.147
4.383-5.019
0.2
29
0.167
243
0.17
272
0.974
0.984
100
0.171
5.019-6.049
0.213
24
0.149
216
0.154
240
0.975
0.987
100
0.148
6.049-8.174
0.296
23
0.164
169
0.177
192
0.955
0.981
100
0.171
8.174-19.606
0.174
6
0.217
136
0.214
142
0.981
0.973
100
0.244
Refinement TLS params.
Method: refined / Origin x: -0.921 Å / Origin y: 20.299 Å / Origin z: 18.436 Å
11
12
13
21
22
23
31
32
33
T
0.0044 Å2
-0.0011 Å2
-0.0235 Å2
-
0.0123 Å2
-0.0068 Å2
-
-
0.2554 Å2
L
2.9919 °2
-1.5091 °2
-0.2878 °2
-
3.2269 °2
-0.3975 °2
-
-
1.7518 °2
S
0.0473 Å °
-0.0595 Å °
-0.1262 Å °
-0.0424 Å °
0.009 Å °
0.4131 Å °
-0.0025 Å °
-0.1174 Å °
-0.0563 Å °
Refinement TLS group
ID
Refine-ID
Refine TLS-ID
Selection
Auth asym-ID
Auth seq-ID
1
X-RAY DIFFRACTION
1
ALL
A
1 - 129
2
X-RAY DIFFRACTION
1
ALL
A
210
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United Kingdom, 1items
United Kingdom, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads







 PDBj
PDBj







 Assembly
Assembly

 Mass: 35.453 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Cl
 Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na
Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na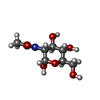
 Type: D-saccharide, alpha linking / Mass: 221.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H15NO6 / Feature type: SUBJECT OF INVESTIGATION
Type: D-saccharide, alpha linking / Mass: 221.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H15NO6 / Feature type: SUBJECT OF INVESTIGATION Mass: 18.015 Da / Num. of mol.: 84 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 84 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing