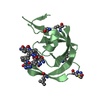
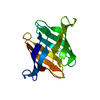
- PDB-2yan: Crystal structure of the second glutaredoxin domain of human TXNL2 -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 2yan
Title
Crystal structure of the second glutaredoxin domain of human TXNL2
Components
GLUTAREDOXIN-3
Keywords
OXIDOREDUCTASE
Function / homology
Function and homology information
negative regulation of cardiac muscle hypertrophy / regulation of the force of heart contraction / iron-sulfur cluster assembly complex / [2Fe-2S] cluster assembly / iron-sulfur cluster assembly / iron-sulfur cluster binding / cell redox homeostasis / protein kinase C binding / Iron uptake and transport / Z disc ...negative regulation of cardiac muscle hypertrophy / regulation of the force of heart contraction / iron-sulfur cluster assembly complex / [2Fe-2S] cluster assembly / iron-sulfur cluster assembly / iron-sulfur cluster binding / cell redox homeostasis / protein kinase C binding / Iron uptake and transport / Z disc / cell cortex / intracellular iron ion homeostasis / dendrite / RNA binding / metal ion binding / identical protein binding / nucleus / cytosol / cytoplasm Similarity search - Function
Resolution: 1.9→19.72 Å / Cor.coef. Fo:Fc: 0.963 / Cor.coef. Fo:Fc free: 0.949 / SU B: 4.093 / SU ML: 0.066 / Cross valid method: THROUGHOUT / ESU R: 0.097 / ESU R Free: 0.098 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.21237
1872
5 %
RANDOM
Rwork
0.18491
-
-
-
obs
0.18629
35624
99.79 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information HOMO SAPIENS (human)
HOMO SAPIENS (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads












 PDBj
PDBj



 Assembly
Assembly







 Mass: 62.068 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 55.845 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe
Mass: 55.845 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
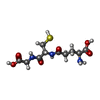
Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 307.323 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H17N3O6S
Mass: 307.323 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H17N3O6S Sample preparation
Sample preparation / Beamline: I02 / Wavelength: 1
/ Beamline: I02 / Wavelength: 1  Processing
Processing