 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2x0k: Crystal structure of modular FAD synthetase from Corynebacterium ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2x0k | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of modular FAD synthetase from Corynebacterium ammoniagenes | ||||||
 Components Components | RIBOFLAVIN BIOSYNTHESIS PROTEIN RIBF | ||||||
 Keywords Keywords | TRANSFERASE / RIBOFLAVIN KINASE / NUCLEOTIDE-BINDING / ATP-BINDING / MULTIFUNCTIONAL ENZYME / NUCLEOTIDYLTRANSFERASE | ||||||
| Function / homology |  Function and homology information Function and homology informationriboflavin kinase / FAD synthase / FMN adenylyltransferase activity / FAD biosynthetic process / riboflavin kinase activity / FMN biosynthetic process / riboflavin biosynthetic process / ATP binding Similarity search - Function | ||||||
| Biological species |  CORYNEBACTERIUM AMMONIAGENES (bacteria) CORYNEBACTERIUM AMMONIAGENES (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.95 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.95 Å | ||||||
 Authors Authors | Herguedas, B. / Hermoso, J.A. / Martinez-Julvez, M. / Medina, M. / Frago, S. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2010 Title: Oligomeric State in the Crystal Structure of Modular Fad Synthetase Provides Insights Into its Sequential Catalysis in Prokaryotes Authors: Herguedas, B. / Martinez-Julvez, M. / Frago, S. / Medina, M. / Hermoso, J.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2x0k.cif.gz | 153.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2x0k.ent.gz | 123.1 KB | Display | PDB format |
| PDBx/mmJSON format | 2x0k.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/x0/2x0kftp://data.pdbj.org/pub/pdb/validation_reports/x0/2x0k | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|






-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 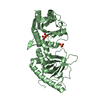
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 36882.359 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) CORYNEBACTERIUM AMMONIAGENES (bacteria)Plasmid: PET28 / Production host: #2: Chemical |   Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#3: Chemical | 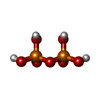  Mass: 177.975 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: H4O7P2 Mass: 177.975 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: H4O7P2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 523 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 523 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.68 Å3/Da / Density % sol: 54.2 % / Description: NONE |
|---|---|
| Crystal grow | pH: 7.5 / Details: 1.5M LI2SO4, 0.1 M HEPES-NAOH, PH 7.5 |
-Data collection
| Diffraction |
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source |
| ||||||||||||||||||
| Detector |
| ||||||||||||||||||
| Radiation |
| ||||||||||||||||||
| Radiation wavelength |
| ||||||||||||||||||
| Reflection | Resolution: 1.95→54.23 Å / Num. obs: 57834 / % possible obs: 100 % / Observed criterion σ(I): 3 / Redundancy: 11.4 % / Rmerge(I) obs: 0.09 / Net I/σ(I): 19.9 | ||||||||||||||||||
| Reflection shell | Resolution: 1.95→2.06 Å / Redundancy: 8.7 % / Rmerge(I) obs: 0.46 / Mean I/σ(I) obs: 2.7 / % possible all: 100 |

- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD Starting model: NONE Resolution: 1.95→94.49 Å / Cor.coef. Fo:Fc: 0.943 / Cor.coef. Fo:Fc free: 0.93 / SU B: 6.813 / SU ML: 0.106 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R: 0.182 / ESU R Free: 0.158 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. ATOM RECORD CONTAINS RESIDUAL B FACTORS ONLY. ALTERNATIVE CONFORMATIONS WITH 0.5 OCCUPANCY HAVE BEEN MODELED FOR RESIDUES A195, B1195, A323- ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. ATOM RECORD CONTAINS RESIDUAL B FACTORS ONLY. ALTERNATIVE CONFORMATIONS WITH 0.5 OCCUPANCY HAVE BEEN MODELED FOR RESIDUES A195, B1195, A323-A331 AND B1321-B1331.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 22.147 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.95→94.49 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|