Mass: 18.015 Da / Num. of mol.: 966 / Source method: isolated from a natural source / Formula: H2O
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.76 Å3/Da / Density % sol: 55.5 % / Description: NONE
Crystal grow
Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 8 Details: CRYSTALS OF THE C. FREUNDII TPL WERE GROWN AT 277 AND 293 K USING THE HANGING DROP VAPOR DIFFUSION METHOD. THE BEST CRYSTALS WERE OBTAINED BY MIXING 2 UL OF THE PROTEIN SOLUTION (18-20 MG/ML) ...Details: CRYSTALS OF THE C. FREUNDII TPL WERE GROWN AT 277 AND 293 K USING THE HANGING DROP VAPOR DIFFUSION METHOD. THE BEST CRYSTALS WERE OBTAINED BY MIXING 2 UL OF THE PROTEIN SOLUTION (18-20 MG/ML) CONTAINING 50MM K-PHOSPHATE PH 8.0, 0.5 MM PLP, 1MM DDT WITH AN EQUAL VOLUME OF THE RESERVOIR SOLUTION CONTAINING 50 MM TRIETHANOLAMINE BUFFER (PH 8.0), 0.5 MM PLP, 2 MM DDT, 0.4 M KCL, AND 35-38% (W/V) POLY(ETHYLENE GLYCOL) 5000 MONOMETHYL ETHER. QUINONOID INTERMEDIATE WITH ALANINE WAS PREPARED BY SOAKING THE TPL CRYSTALS FOR 5 MIN IN THE STABILIZATION SOLUTION CONTAINING 40% POLY(ETHYLENE GLYCOL) 5000 MONOMETHYL ETHER, 50 MM TRIETHANOLAMINE BUFFER (PH 8.0), 0.25 M KCL, 0.2 MM PLP, 0.5 MM DTT AND 10 MM L-ALA.
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information CITROBACTER FREUNDII (bacteria)
CITROBACTER FREUNDII (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads



















 PDBj
PDBj Assembly
Assembly

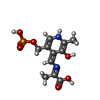




 Mass: 318.220 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C11H15N2O7P
Mass: 318.220 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C11H15N2O7P Mass: 150.173 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O4
Mass: 150.173 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O4 Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K
Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K Mass: 326.383 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H30O8 / Comment: precipitant*YM
Mass: 326.383 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H30O8 / Comment: precipitant*YM Sample preparation
Sample preparation / Beamline: BW7B / Wavelength: 0.862
/ Beamline: BW7B / Wavelength: 0.862  Processing
Processing