 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2q6f: Crystal structure of infectious bronchitis virus (IBV) main prote... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2q6f | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of infectious bronchitis virus (IBV) main protease in complex with a Michael acceptor inhibitor N3 | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | HYDROLASE / coronavirus / IBV / main protease / 3C-Like proteinase / Michael acceptor inhibitor | |||||||||
| Function / homology |  Function and homology information Function and homology informationhost cell membrane / viral genome replication / peptidase activity / transferase activity / omega peptidase activity / ubiquitinyl hydrolase 1 / Hydrolases; Acting on peptide bonds (peptidases); Cysteine endopeptidases / lyase activity / cysteine-type deubiquitinase activity / viral protein processing ...host cell membrane / viral genome replication / peptidase activity / transferase activity / omega peptidase activity / ubiquitinyl hydrolase 1 / Hydrolases; Acting on peptide bonds (peptidases); Cysteine endopeptidases / lyase activity / cysteine-type deubiquitinase activity / viral protein processing / host cell perinuclear region of cytoplasm / host cell endoplasmic reticulum membrane / viral translational frameshifting / symbiont-mediated activation of host autophagy / cysteine-type endopeptidase activity / proteolysis / RNA binding / zinc ion binding / metal ion binding Similarity search - Function | |||||||||
| Biological species |  Infectious bronchitis virus Infectious bronchitis virussynthetic construct (others) | |||||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å | |||||||||
 Authors Authors | Xue, X.Y. / Yang, H.T. / Xue, F. / Bartlam, M. / Rao, Z.H. | |||||||||
 Citation Citation | Journal: J.Virol. / Year: 2008 Title: Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. Authors: Xue, X. / Yu, H. / Yang, H. / Xue, F. / Wu, Z. / Shen, W. / Li, J. / Zhou, Z. / Ding, Y. / Zhao, Q. / Zhang, X.C. / Liao, M. / Bartlam, M. / Rao, Z. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2q6f.cif.gz | 137.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2q6f.ent.gz | 106.2 KB | Display | PDB format |
| PDBx/mmJSON format | 2q6f.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/q6/2q6fftp://data.pdbj.org/pub/pdb/validation_reports/q6/2q6f | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2q6dSC  2q6gC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological assembly is a dimer with one inhibitor molecule in the active site of each protomer. |
-Components
| #1: Protein | Mass: 33635.949 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Infectious bronchitis virus / Genus: Coronavirus / Strain: M41 / Gene: M41 3C-like protease gene / Plasmid: pGEX-4T-1 / Species (production host): Escherichia coli / Production host:  References: UniProt: Q3Y5H1, UniProt: P0C6V5*PLUS, Hydrolases; Acting on peptide bonds (peptidases); Cysteine endopeptidases #2: Protein/peptide | 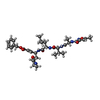  Type: Peptide-like / Class: Inhibitor / Mass: 680.791 Da / Num. of mol.: 2 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) Type: Peptide-like / Class: Inhibitor / Mass: 680.791 Da / Num. of mol.: 2 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others)References: N-[(5-METHYLISOXAZOL-3-YL)CARBONYL]ALANYL-L-VALYL-N~1~-((1R,2Z)-4-(BENZYLOXY)-4-OXO-1-{[(3R)-2-OXOPYRROLIDIN-3-YL]METHYL}BUT-2-ENYL)-L-LEUCINAMIDE #3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 419 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 419 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.58 Å3/Da / Density % sol: 52.29 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: 20% PEG10000, 0.1M HEPES pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 / Wavelength: 1.5418 Å |
| Detector | Type: RIGAKU RAXIS IV++ / Detector: IMAGE PLATE / Date: Dec 8, 2006 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2→50 Å / Num. obs: 42883 / % possible obs: 94.2 % / Observed criterion σ(I): 0 / Redundancy: 3.9 % / Rmerge(I) obs: 0.041 |
| Reflection shell | Resolution: 2→2.07 Å / Redundancy: 3.3 % / Rmerge(I) obs: 0.225 / Mean I/σ(I) obs: 5.1 / Num. unique all: 3787 / % possible all: 82.6 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2Q6D Resolution: 2→50 Å / Isotropic thermal model: Isotropic / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→50 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
|
