 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2j5t: Glutamate 5-kinase from Escherichia coli complexed with glutamate -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2j5t | ||||||
|---|---|---|---|---|---|---|---|
| Title | Glutamate 5-kinase from Escherichia coli complexed with glutamate | ||||||
 Components Components | GLUTAMATE 5-KINASE | ||||||
 Keywords Keywords | TRANSFERASE / PROLINE BIOSYNTHESIS / FEEDBACK REGULATION | ||||||
| Function / homology |  Function and homology information Function and homology informationproline binding / glutamate 5-kinase / glutamate 5-kinase activity / L-proline biosynthetic process / magnesium ion binding / protein homodimerization activity / RNA binding / ATP binding / identical protein binding / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.9 Å | ||||||
 Authors Authors | Marco-Marin, C. / Gil-Ortiz, F. / Perez-Arellano, I. / Cervera, J. / Fita, I. / Rubio, V. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2007 Title: A Novel Two-Domain Architecture within the Amino Acid Kinase Enzyme Family Revealed by the Crystal Structure of Escherichia Coli Glutamate 5-Kinase. Authors: Marco-Marin, C. / Gil-Ortiz, F. / Perez-Arellano, I. / Cervera, J. / Fita, I. / Rubio, V. | ||||||
| History |
| ||||||
| Remark 650 | HELIX DETERMINATION METHOD: AUTHOR PROVIDED. | ||||||
| Remark 700 | SHEET DETERMINATION METHOD: AUTHOR PROVIDED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2j5t.cif.gz | 518.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2j5t.ent.gz | 427.8 KB | Display | PDB format |
| PDBx/mmJSON format | 2j5t.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/j5/2j5tftp://data.pdbj.org/pub/pdb/validation_reports/j5/2j5t | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2j5vSC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: 1 / Ens-ID: 1 / Beg auth comp-ID: ASP / Beg label comp-ID: ASP / End auth comp-ID: ARG / End label comp-ID: ARG / Refine code: 4 / Auth seq-ID: 3 - 367 / Label seq-ID: 3 - 367
NCS oper:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | CONTACTS BETWEEN DIMERS ARE WEAK, BUT GEL FILTRATION EXPERIMENTS SHOW THAT THE PROTEIN IS TETRAMERIC AND THE TETRAMERS FORMED IN THE CRYSTAL ARE EQUIVALENT TO THOSE FOUNDIN A DIFFERENT CRYSTAL OF THIS PROTEIN (WITH DIFFERENT SPACEGROUP). |
-Components
-Protein , 1 types, 8 molecules ABCDEFGH
| #1: Protein | Mass: 39089.430 Da / Num. of mol.: 8 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|
-Non-polymers , 5 types, 151 molecules 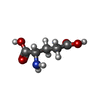




| #2: Chemical | ChemComp-GLU /  Type: L-peptide linking / Mass: 147.129 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: C5H9NO4 Type: L-peptide linking / Mass: 147.129 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: C5H9NO4#3: Chemical | ChemComp-SO4 /  Mass: 96.063 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: SO4#4: Chemical | ChemComp-CL /  Mass: 35.453 Da / Num. of mol.: 20 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 20 / Source method: obtained synthetically / Formula: Cl#5: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Mg#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 103 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Compound details | ENGINEERED RESIDUE IN CHAIN A, ILE 129 TO VAL ENGINEERED RESIDUE IN CHAIN B, ILE 129 TO VAL ...ENGINEERED |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.77 Å3/Da / Density % sol: 55.2 % |
|---|---|
| Crystal grow | pH: 6.5 / Details: 1.45 M MGSO4, 20 MM CACL2, 0.1 M MES PH 6.5. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-4 / Wavelength: 0.979 / Beamline: ID14-4 / Wavelength: 0.979 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Feb 6, 2005 / Details: TOROIDAL MIRRORS |
| Radiation | Monochromator: SI(111) AND SI (311) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.979 Å / Relative weight: 1 |
| Reflection | Resolution: 2.9→50 Å / Num. obs: 74908 / % possible obs: 99.3 % / Observed criterion σ(I): 2.9 / Redundancy: 4.5 % / Rmerge(I) obs: 0.16 / Net I/σ(I): 9.7 |
| Reflection shell | Resolution: 2.9→3.06 Å / Redundancy: 4.6 % / Rmerge(I) obs: 0.49 / Mean I/σ(I) obs: 2.9 / % possible all: 99.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PARTIALLY-REFINED MODEL OF PBD ENTRY 2J5V Resolution: 2.9→25 Å / Cor.coef. Fo:Fc: 0.92 / Cor.coef. Fo:Fc free: 0.874 / SU B: 35.798 / SU ML: 0.302 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R Free: 0.401 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. THE FINAL STRUCTURE ENCOMPASSES THE ENTIRE POLYPEPTIDE CHAIN FROM RESIDUE 3 IN SUBUNITS D AND E, BUT LACKS THE FOLLOWING RESIDUES IN THE ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. THE FINAL STRUCTURE ENCOMPASSES THE ENTIRE POLYPEPTIDE CHAIN FROM RESIDUE 3 IN SUBUNITS D AND E, BUT LACKS THE FOLLOWING RESIDUES IN THE OTHER SUBUNITS, 202-211 (A), 202-213 AND 367 (B), 203-211 (G AND C), 176-185 AND 196- 213 (H), 203-213 (F).
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 24.57 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.9→25 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|