exocrine system development / regulation protein catabolic process at presynapse / regulated exocytosis / regulation of exocytosis / RAB geranylgeranylation / Golgi to plasma membrane protein transport / GMP binding / secretory granule membrane / small monomeric GTPase / synaptic vesicle membrane ...exocrine system development / regulation protein catabolic process at presynapse / regulated exocytosis / regulation of exocytosis / RAB geranylgeranylation / Golgi to plasma membrane protein transport / GMP binding / secretory granule membrane / small monomeric GTPase / synaptic vesicle membrane / endosome / Golgi membrane / GTPase activity / GTP binding / Golgi apparatus / plasma membrane Similarity search - Function
: / ARF-like small GTPases; ARF, ADP-ribosylation factor / Small GTPase Rab domain profile. / Ran (Ras-related nuclear proteins) /TC4 subfamily of small GTPases / Rho (Ras homology) subfamily of Ras-like small GTPases / Ras subfamily of RAS small GTPases / Small GTPase / Ras family / Rab subfamily of small GTPases / Small GTP-binding protein domain ...: / ARF-like small GTPases; ARF, ADP-ribosylation factor / Small GTPase Rab domain profile. / Ran (Ras-related nuclear proteins) /TC4 subfamily of small GTPases / Rho (Ras homology) subfamily of Ras-like small GTPases / Ras subfamily of RAS small GTPases / Small GTPase / Ras family / Rab subfamily of small GTPases / Small GTP-binding protein domain / P-loop containing nucleotide triphosphate hydrolases / Rossmann fold / P-loop containing nucleoside triphosphate hydrolase / 3-Layer(aba) Sandwich / Alpha Beta Similarity search - Domain/homology
BIOMOLECULE: 1 THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT WHICH CONSISTS OF 1 CHAIN(S) ...BIOMOLECULE: 1 THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT WHICH CONSISTS OF 1 CHAIN(S). THE BIOLOGICAL MOLECULE FOR THE PROTEIN IS UNKNOWN.
Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 1.5418 Å / Relative weight: 1
Reflection
Resolution: 2→30 Å / Num. obs: 12581 / % possible obs: 99.5 % / Redundancy: 7.6 % / Rmerge(I) obs: 0.173 / Χ2: 1.017 / Net I/σ(I): 4.4
Reflection shell
Resolution (Å)
% possible obs (%)
Redundancy (%)
Rmerge(I) obs
Num. unique obs
Χ2
2-2.07
95.7
6.3
0.841
1178
0.863
2.07-2.15
99.3
7.2
0.716
1224
0.866
2.15-2.25
100
7.7
0.603
1240
0.923
2.25-2.37
99.9
7.8
0.495
1231
0.903
2.37-2.52
100
7.8
0.44
1253
0.927
2.52-2.71
100
7.9
0.358
1250
0.909
2.71-2.99
100
7.9
0.235
1259
1.042
2.99-3.42
100
7.9
0.139
1277
1.122
3.42-4.31
100
7.8
0.088
1286
1.274
4.31-30
100
7.4
0.068
1383
1.262
-
Phasing
Phasing
Method: molecular replacement
Phasing MR
Highest resolution
Lowest resolution
Rotation
2.5 Å
19.58 Å
Translation
2.5 Å
19.58 Å
-
Processing
Software
Name
Version
Classification
NB
DENZO
datareduction
SCALEPACK
datascaling
PHASER
phasing
REFMAC
refmac_5.2.0019
refinement
PDB_EXTRACT
1.701
dataextraction
Refinement
Method to determine structure: MOLECULAR REPLACEMENT Starting model: pdb entry 1G16_A Resolution: 2→30 Å / Cor.coef. Fo:Fc: 0.936 / Cor.coef. Fo:Fc free: 0.888 / WRfactor Rfree: 0.236 / WRfactor Rwork: 0.181 / SU B: 4.67 / SU ML: 0.133 / ESU R: 0.198 / ESU R Free: 0.192 Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. ARP/wARP was also used for the refinement.
Rfactor
Num. reflection
% reflection
Rfree
0.2769
609
4.876 %
Rwork
0.2096
-
-
all
0.213
-
-
obs
-
12491
99.673 %
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK BULK SOLVENT
Displacement parameters
Biso mean: 19.721 Å2
Baniso -1
Baniso -2
Baniso -3
1-
0.059 Å2
0 Å2
0 Å2
2-
-
0.615 Å2
0 Å2
3-
-
-
-0.674 Å2
Refinement step
Cycle: LAST / Resolution: 2→30 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
1272
0
37
62
1371
Refine LS restraints
Refine-ID
Type
Dev ideal
Dev ideal target
Number
X-RAY DIFFRACTION
r_bond_refined_d
0.018
0.022
1331
X-RAY DIFFRACTION
r_bond_other_d
0.002
0.02
865
X-RAY DIFFRACTION
r_angle_refined_deg
1.461
1.984
1810
X-RAY DIFFRACTION
r_angle_other_deg
1.018
3
2117
X-RAY DIFFRACTION
r_dihedral_angle_1_deg
5.4
5
167
X-RAY DIFFRACTION
r_dihedral_angle_2_deg
34.279
24.386
57
X-RAY DIFFRACTION
r_dihedral_angle_3_deg
13.782
15
226
X-RAY DIFFRACTION
r_dihedral_angle_4_deg
17.854
15
6
X-RAY DIFFRACTION
r_chiral_restr
0.08
0.2
206
X-RAY DIFFRACTION
r_gen_planes_refined
0.006
0.02
1469
X-RAY DIFFRACTION
r_gen_planes_other
0.003
0.02
271
X-RAY DIFFRACTION
r_nbd_refined
0.2
0.3
261
X-RAY DIFFRACTION
r_nbd_other
0.209
0.3
890
X-RAY DIFFRACTION
r_nbtor_refined
0.177
0.5
612
X-RAY DIFFRACTION
r_nbtor_other
0.087
0.5
672
X-RAY DIFFRACTION
r_xyhbond_nbd_refined
0.167
0.5
123
X-RAY DIFFRACTION
r_metal_ion_refined
0.027
0.5
1
X-RAY DIFFRACTION
r_symmetry_vdw_refined
0.202
0.3
5
X-RAY DIFFRACTION
r_symmetry_vdw_other
0.31
0.3
16
X-RAY DIFFRACTION
r_symmetry_hbond_refined
0.311
0.5
7
X-RAY DIFFRACTION
r_mcbond_it
2.367
2
839
X-RAY DIFFRACTION
r_mcbond_other
0.558
2
340
X-RAY DIFFRACTION
r_mcangle_it
3.22
3
1291
X-RAY DIFFRACTION
r_scbond_it
2.342
2
583
X-RAY DIFFRACTION
r_scangle_it
3.249
3
516
LS refinement shell
Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Rfactor all
Num. reflection all
% reflection obs (%)
2-2.052
0.408
42
0.272
849
0.278
917
97.165
2.052-2.108
0.356
53
0.239
811
0.246
865
99.884
2.108-2.169
0.382
44
0.233
809
0.24
864
98.727
2.169-2.235
0.281
36
0.217
787
0.22
823
100
2.235-2.308
0.261
43
0.204
780
0.208
824
99.879
2.308-2.388
0.247
38
0.204
734
0.207
772
100
2.388-2.478
0.251
38
0.208
721
0.21
759
100
2.478-2.578
0.298
34
0.216
693
0.22
729
99.726
2.578-2.692
0.244
35
0.223
673
0.224
708
100
2.692-2.822
0.297
29
0.224
648
0.227
677
100
2.822-2.973
0.244
30
0.226
624
0.227
654
100
2.973-3.152
0.253
30
0.221
579
0.223
609
100
3.152-3.367
0.246
21
0.202
551
0.204
572
100
3.367-3.633
0.334
29
0.185
519
0.191
548
100
3.633-3.974
0.28
24
0.192
472
0.196
496
100
3.974-4.433
0.244
26
0.167
436
0.172
462
100
4.433-5.1
0.236
17
0.163
402
0.166
419
100
5.1-6.202
0.349
12
0.248
339
0.251
351
100
6.202-8.589
0.317
21
0.246
270
0.251
291
100
8.589-30
0.128
7
0.232
185
0.227
192
100
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads





 PDBj
PDBj



 Assembly
Assembly


 Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg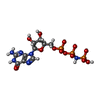
 Mass: 522.196 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N6O13P3
Mass: 522.196 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N6O13P3
 Num. of mol.: 4 / Source method: obtained synthetically
Num. of mol.: 4 / Source method: obtained synthetically Mass: 18.015 Da / Num. of mol.: 62 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 62 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing