- PDB-2fme: Crystal structure of the mitotic kinesin eg5 (ksp) in complex wit... -
+
データを開く
IDまたはキーワード:
読み込み中...
-
基本情報
登録情報
データベース: PDB / ID: 2fme
タイトル
Crystal structure of the mitotic kinesin eg5 (ksp) in complex with mg-adp and (r)-4-(3-hydroxyphenyl)-n,n,7,8-tetramethyl-3,4-dihydroisoquinoline-2(1h)-carboxamide
要素
Kinesin-like protein KIF11
キーワード
CELL CYCLE / Eg5 KSP Mg-ADP Complex Inhibitor
機能・相同性
機能・相同性情報
spindle elongation / regulation of mitotic centrosome separation / plus-end-directed microtubule motor activity / mitotic centrosome separation / Kinesins / spindle organization / kinesin complex / microtubule motor activity / COPI-dependent Golgi-to-ER retrograde traffic / microtubule-based movement ...spindle elongation / regulation of mitotic centrosome separation / plus-end-directed microtubule motor activity / mitotic centrosome separation / Kinesins / spindle organization / kinesin complex / microtubule motor activity / COPI-dependent Golgi-to-ER retrograde traffic / microtubule-based movement / mitotic spindle assembly / MHC class II antigen presentation / mitotic spindle organization / sperm end piece / spindle / spindle pole / mitotic spindle / mitotic cell cycle / sperm principal piece / sperm midpiece / microtubule binding / microtubule / ciliary basal body / cell division / protein kinase binding / protein-containing complex / ATP binding / membrane / nucleus / cytosol 類似検索 - 分子機能
Kinesin-associated microtubule-binding domain / Kinesin-associated microtubule-binding / : / : / Kinesin motor domain / Kinesin / Kinesin motor domain signature. / Kinesin motor domain, conserved site / Kinesin motor domain / Kinesin motor domain profile. ...Kinesin-associated microtubule-binding domain / Kinesin-associated microtubule-binding / : / : / Kinesin motor domain / Kinesin / Kinesin motor domain signature. / Kinesin motor domain, conserved site / Kinesin motor domain / Kinesin motor domain profile. / Kinesin motor, catalytic domain. ATPase. / Kinesin motor domain / Kinesin motor domain superfamily / P-loop containing nucleoside triphosphate hydrolase / 3-Layer(aba) Sandwich / Alpha Beta 類似検索 - ドメイン・相同性
Chem-3QC / ADENOSINE-5'-DIPHOSPHATE / Kinesin-like protein KIF11 類似検索 - 構成要素
Kinesin-likeproteinKIF11 / Kinesin-related motor protein Eg5 / Kinesin-like spindle protein HKSP / Thyroid receptor ...Kinesin-related motor protein Eg5 / Kinesin-like spindle protein HKSP / Thyroid receptor interacting protein 5 / TRIP5 / Kinesin-like protein 1
温度: 277 K / pH: 7.8 詳細: 1:1 reservoir: 0.1 M NaHEPES pH 7.8, 0.2 M MgCl2, 23% PEG-3350; protein: 25 mg/ml (610 uM) in 25 mM PIPES pH 6.8, 200 mM NaCl, 2 mM DTT, 5 mM MgCl2, 1 mM EGTA, 2.5 mM ADP, 0.75% DMSO, uM (R)- ...詳細: 1:1 reservoir: 0.1 M NaHEPES pH 7.8, 0.2 M MgCl2, 23% PEG-3350; protein: 25 mg/ml (610 uM) in 25 mM PIPES pH 6.8, 200 mM NaCl, 2 mM DTT, 5 mM MgCl2, 1 mM EGTA, 2.5 mM ADP, 0.75% DMSO, uM (R)-4-(3-HYDROXYPHENYL)-N,N,7,8-TETRAMETHYL-3,4-DIHYDROISOQUINOLINE-2(1H)-CARBOXAMIDE, vapor diffusion, hanging drop, temperature 277K, pH 7.80
手法: Solvent flattening and Histogram matching / 反射: 47350
Phasing dm shell
解像度 (Å)
Delta phi final
FOM
反射
9.79-100
27.1
0.768
505
7.42-9.79
30.8
0.882
659
6.22-7.42
37.2
0.824
815
5.46-6.22
33.1
0.863
961
4.92-5.46
25
0.892
1040
4.52-4.92
22.3
0.917
1159
4.2-4.52
22.5
0.918
1227
3.94-4.2
23.4
0.889
1336
3.72-3.94
20.4
0.91
1379
3.54-3.72
19.4
0.91
1473
3.38-3.54
21.9
0.905
1539
3.24-3.38
23.2
0.897
1605
3.12-3.24
23.2
0.878
1653
3-3.12
25.7
0.855
1718
2.91-3
26.1
0.835
1773
2.81-2.91
26.4
0.841
1813
2.73-2.81
26.7
0.836
1897
2.66-2.73
26.3
0.841
1931
2.59-2.66
26.1
0.85
1988
2.52-2.59
25
0.855
2001
2.46-2.52
25.9
0.843
2085
2.41-2.46
26.1
0.851
2087
2.36-2.41
24.6
0.845
2149
2.31-2.36
23.6
0.842
2224
2.26-2.31
22.8
0.857
2221
2.22-2.26
24.3
0.849
2254
2.18-2.22
26
0.846
2309
2.14-2.18
28.1
0.848
1961
2.1-2.14
33.2
0.806
1588
-
解析
ソフトウェア
名称
バージョン
分類
NB
DENZO
データ削減
SCALEPACK
データスケーリング
AMoRE
位相決定
直接法
位相決定
CNS
精密化
PDB_EXTRACT
1.701
データ抽出
CNX
2002
精密化
精密化
構造決定の手法: 分子置換 開始モデル: INTERNAL MODEL OF EG5/LIGAND COMPLEX 解像度: 2.1→40 Å / Rfactor Rfree error: 0.01 / Data cutoff high absF: 2140759 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / 交差検証法: THROUGHOUT / σ(F): 0 詳細: Due to a feature in the refinement program, the structure was refined with an OXT on a residue that is not the terminal residue of the sequence. The OXT was changed to N of the next residue ...詳細: Due to a feature in the refinement program, the structure was refined with an OXT on a residue that is not the terminal residue of the sequence. The OXT was changed to N of the next residue by the wwPDB annotation staff.
Rfactor
反射数
%反射
Selection details
Rfree
0.305
944
2 %
RANDOM
Rwork
0.28
-
-
-
obs
-
47350
93.3 %
-
溶媒の処理
溶媒モデル: BABINET / Bsol: 280 Å2 / ksol: 0.9 e/Å3
原子変位パラメータ
Biso mean: 26.9 Å2
Baniso -1
Baniso -2
Baniso -3
1-
2.75 Å2
0 Å2
-1.7 Å2
2-
-
-6.62 Å2
0 Å2
3-
-
-
3.87 Å2
Refine analyze
Free
Obs
Luzzati coordinate error
0.35 Å
0.31 Å
Luzzati d res low
-
5 Å
Luzzati sigma a
0.17 Å
0.16 Å
精密化ステップ
サイクル: LAST / 解像度: 2.1→40 Å
タンパク質
核酸
リガンド
溶媒
全体
原子数
5000
0
104
178
5282
拘束条件
Refine-ID
タイプ
Dev ideal
Dev ideal target
X-RAY DIFFRACTION
c_bond_d
0.008
X-RAY DIFFRACTION
c_bond_d_na
X-RAY DIFFRACTION
c_bond_d_prot
X-RAY DIFFRACTION
c_angle_d
X-RAY DIFFRACTION
c_angle_d_na
X-RAY DIFFRACTION
c_angle_d_prot
X-RAY DIFFRACTION
c_angle_deg
1.5
X-RAY DIFFRACTION
c_angle_deg_na
X-RAY DIFFRACTION
c_angle_deg_prot
X-RAY DIFFRACTION
c_dihedral_angle_d
24.5
X-RAY DIFFRACTION
c_dihedral_angle_d_na
X-RAY DIFFRACTION
c_dihedral_angle_d_prot
X-RAY DIFFRACTION
c_improper_angle_d
0.91
X-RAY DIFFRACTION
c_improper_angle_d_na
X-RAY DIFFRACTION
c_improper_angle_d_prot
X-RAY DIFFRACTION
c_mcbond_it
1.2
1.5
X-RAY DIFFRACTION
c_mcangle_it
1.88
2
X-RAY DIFFRACTION
c_scbond_it
1.73
2
X-RAY DIFFRACTION
c_scangle_it
2.43
2.5
Refine LS restraints NCS
Refine-ID: X-RAY DIFFRACTION / Rms dev Biso: 2 Å2 / Rms dev position: 0.036 Å / Weight Biso: 2 / Weight position: 300
Ens-ID
Dom-ID
NCS model details
1
1
RESTRAINTS
2
2
LS精密化 シェル
解像度: 2.1→2.2 Å / Rfactor Rfree error: 0.034 / Total num. of bins used: 8
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Homo sapiens (ヒト)
Homo sapiens (ヒト) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード









 PDBj
PDBj












 集合体
集合体




 分子量: 24.305 Da / 分子数: 2 / 由来タイプ: 合成 / 式: Mg
分子量: 24.305 Da / 分子数: 2 / 由来タイプ: 合成 / 式: Mg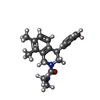
 分子量: 324.417 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C20H24N2O2
分子量: 324.417 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C20H24N2O2
 分子量: 427.201 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C10H15N5O10P2 / コメント: ADP, エネルギー貯蔵分子*YM
分子量: 427.201 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C10H15N5O10P2 / コメント: ADP, エネルギー貯蔵分子*YM 分子量: 18.015 Da / 分子数: 178 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 178 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 解析
解析