- PDB-2fme: Crystal structure of the mitotic kinesin eg5 (ksp) in complex wit... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 2fme
Title
Crystal structure of the mitotic kinesin eg5 (ksp) in complex with mg-adp and (r)-4-(3-hydroxyphenyl)-n,n,7,8-tetramethyl-3,4-dihydroisoquinoline-2(1h)-carboxamide
Components
Kinesin-like protein KIF11
Keywords
CELL CYCLE / Eg5 KSP Mg-ADP Complex Inhibitor
Function / homology
Function and homology information
spindle elongation / regulation of mitotic centrosome separation / plus-end-directed microtubule motor activity / mitotic centrosome separation / Kinesins / spindle organization / microtubule motor activity / kinesin complex / COPI-dependent Golgi-to-ER retrograde traffic / microtubule-based movement ...spindle elongation / regulation of mitotic centrosome separation / plus-end-directed microtubule motor activity / mitotic centrosome separation / Kinesins / spindle organization / microtubule motor activity / kinesin complex / COPI-dependent Golgi-to-ER retrograde traffic / microtubule-based movement / mitotic spindle assembly / MHC class II antigen presentation / mitotic spindle organization / sperm end piece / spindle / spindle pole / mitotic spindle / mitotic cell cycle / sperm principal piece / sperm midpiece / microtubule binding / microtubule / ciliary basal body / cell division / protein kinase binding / protein-containing complex / ATP binding / membrane / nucleus / cytosol Similarity search - Function
Kinesin-associated microtubule-binding domain / Kinesin-associated microtubule-binding / : / : / Kinesin motor domain / Kinesin / Kinesin motor domain signature. / Kinesin motor domain, conserved site / Kinesin motor domain / Kinesin motor domain profile. ...Kinesin-associated microtubule-binding domain / Kinesin-associated microtubule-binding / : / : / Kinesin motor domain / Kinesin / Kinesin motor domain signature. / Kinesin motor domain, conserved site / Kinesin motor domain / Kinesin motor domain profile. / Kinesin motor, catalytic domain. ATPase. / Kinesin motor domain / Kinesin motor domain superfamily / P-loop containing nucleoside triphosphate hydrolase / 3-Layer(aba) Sandwich / Alpha Beta Similarity search - Domain/homology
Kinesin-likeproteinKIF11 / Kinesin-related motor protein Eg5 / Kinesin-like spindle protein HKSP / Thyroid receptor ...Kinesin-related motor protein Eg5 / Kinesin-like spindle protein HKSP / Thyroid receptor interacting protein 5 / TRIP5 / Kinesin-like protein 1
Mass: 41055.582 Da / Num. of mol.: 2 / Fragment: Kinesin-motor domain (RESIDUES 1-368) Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: KIF11, EG5, KNSL1 / Plasmid: pET28N / Species (production host): Escherichia coli / Production host: Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21(DE3) / References: UniProt: P52732
Mass: 18.015 Da / Num. of mol.: 178 / Source method: isolated from a natural source / Formula: H2O
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.68 Å3/Da / Density % sol: 54.02 %
Crystal grow
Temperature: 277 K / pH: 7.8 Details: 1:1 reservoir: 0.1 M NaHEPES pH 7.8, 0.2 M MgCl2, 23% PEG-3350; protein: 25 mg/ml (610 uM) in 25 mM PIPES pH 6.8, 200 mM NaCl, 2 mM DTT, 5 mM MgCl2, 1 mM EGTA, 2.5 mM ADP, 0.75% DMSO, uM (R)- ...Details: 1:1 reservoir: 0.1 M NaHEPES pH 7.8, 0.2 M MgCl2, 23% PEG-3350; protein: 25 mg/ml (610 uM) in 25 mM PIPES pH 6.8, 200 mM NaCl, 2 mM DTT, 5 mM MgCl2, 1 mM EGTA, 2.5 mM ADP, 0.75% DMSO, uM (R)-4-(3-HYDROXYPHENYL)-N,N,7,8-TETRAMETHYL-3,4-DIHYDROISOQUINOLINE-2(1H)-CARBOXAMIDE, vapor diffusion, hanging drop, temperature 277K, pH 7.80
Method: Solvent flattening and Histogram matching / Reflection: 47350
Phasing dm shell
Resolution (Å)
Delta phi final
FOM
Reflection
9.79-100
27.1
0.768
505
7.42-9.79
30.8
0.882
659
6.22-7.42
37.2
0.824
815
5.46-6.22
33.1
0.863
961
4.92-5.46
25
0.892
1040
4.52-4.92
22.3
0.917
1159
4.2-4.52
22.5
0.918
1227
3.94-4.2
23.4
0.889
1336
3.72-3.94
20.4
0.91
1379
3.54-3.72
19.4
0.91
1473
3.38-3.54
21.9
0.905
1539
3.24-3.38
23.2
0.897
1605
3.12-3.24
23.2
0.878
1653
3-3.12
25.7
0.855
1718
2.91-3
26.1
0.835
1773
2.81-2.91
26.4
0.841
1813
2.73-2.81
26.7
0.836
1897
2.66-2.73
26.3
0.841
1931
2.59-2.66
26.1
0.85
1988
2.52-2.59
25
0.855
2001
2.46-2.52
25.9
0.843
2085
2.41-2.46
26.1
0.851
2087
2.36-2.41
24.6
0.845
2149
2.31-2.36
23.6
0.842
2224
2.26-2.31
22.8
0.857
2221
2.22-2.26
24.3
0.849
2254
2.18-2.22
26
0.846
2309
2.14-2.18
28.1
0.848
1961
2.1-2.14
33.2
0.806
1588
-
Processing
Software
Name
Version
Classification
NB
DENZO
datareduction
SCALEPACK
datascaling
AMoRE
phasing
DM
phasing
CNS
refinement
PDB_EXTRACT
1.701
dataextraction
CNX
2002
refinement
Refinement
Method to determine structure: MOLECULAR REPLACEMENT Starting model: INTERNAL MODEL OF EG5/LIGAND COMPLEX Resolution: 2.1→40 Å / Rfactor Rfree error: 0.01 / Data cutoff high absF: 2140759 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: Due to a feature in the refinement program, the structure was refined with an OXT on a residue that is not the terminal residue of the sequence. The OXT was changed to N of the next residue ...Details: Due to a feature in the refinement program, the structure was refined with an OXT on a residue that is not the terminal residue of the sequence. The OXT was changed to N of the next residue by the wwPDB annotation staff.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj












 Assembly
Assembly




 Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg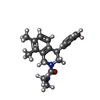
 Mass: 324.417 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H24N2O2
Mass: 324.417 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H24N2O2
 Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM
Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 18.015 Da / Num. of mol.: 178 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 178 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing