 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2c5w: PENICILLIN-BINDING PROTEIN 1A (PBP-1A) ACYL-ENZYME COMPLEX (CEFOT... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2c5w | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | PENICILLIN-BINDING PROTEIN 1A (PBP-1A) ACYL-ENZYME COMPLEX (CEFOTAXIME) FROM STREPTOCOCCUS PNEUMONIAE | |||||||||
 Components Components | (PENICILLIN-BINDING PROTEIN ...) x 2 | |||||||||
 Keywords Keywords | TRANSFERASE/HYDROLASE / PEPTIDOGLYCAN SYNTHESIS / TRANSFERASE-HYDROLASE COMPLEX / ANTIBIOTIC RESISTANCE / CELL SHAPE / MULTIFUNCTIONAL ENZYME | |||||||||
| Function / homology |  Function and homology information Function and homology informationpeptidoglycan glycosyltransferase / peptidoglycan glycosyltransferase activity / serine-type D-Ala-D-Ala carboxypeptidase / serine-type D-Ala-D-Ala carboxypeptidase activity / penicillin binding / peptidoglycan biosynthetic process / cell wall organization / regulation of cell shape / outer membrane-bounded periplasmic space / response to antibiotic ...peptidoglycan glycosyltransferase / peptidoglycan glycosyltransferase activity / serine-type D-Ala-D-Ala carboxypeptidase / serine-type D-Ala-D-Ala carboxypeptidase activity / penicillin binding / peptidoglycan biosynthetic process / cell wall organization / regulation of cell shape / outer membrane-bounded periplasmic space / response to antibiotic / proteolysis / extracellular region Similarity search - Function | |||||||||
| Biological species |   STREPTOCOCCUS PNEUMONIAE (bacteria) STREPTOCOCCUS PNEUMONIAE (bacteria) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.55 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.55 Å | |||||||||
 Authors Authors | Contreras-Martel, C. / Job, V. / Di Guilmi, A.-M. / Vernet, T. / Dideberg, O. / Dessen, A. | |||||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2006 Title: Crystal Structure of Penicillin-Binding Protein 1A (Pbp1A) Reveals a Mutational Hotspot Implicated in Beta-Lactam Resistance in Streptococcus Pneumoniae. Authors: Contreras-Martel, C. / Job, V. / Di Guilmi, A.-M. / Vernet, T. / Dideberg, O. / Dessen, A. #1: Journal: Acta Crystallogr.,Sect.D / Year: 2003 Title: Structural Studies of the Transpeptidase Domain of Pbp1A from Streptococcus Pneumoniae Authors: Job, V. / Di Guilmi, A.-M. / Martin, L. / Vernet, T. / Dideberg, O. / Dessen, A. #2: Journal: J.Bacteriol. / Year: 1998 Title: Identification, Purification, and Charactherization of Transpeptidase and Glycosyltransferase Domains of Streptococcus Pneumoniae Penicillin-Binding Protein 1A Authors: Di Guilmi, A.-M. / Mouz, N. / Andrieu, J.-P. / Hoskins, J. / Jaskunas, S.R. / Gagnon, J. / Dideberg, O. / Vernet, T. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2c5w.cif.gz | 96.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2c5w.ent.gz | 72.7 KB | Display | PDB format |
| PDBx/mmJSON format | 2c5w.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/c5/2c5wftp://data.pdbj.org/pub/pdb/validation_reports/c5/2c5w | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2c6wC  2bg4 C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | PBP-1A IS A MONOMER IN SOLUTION, BUT SINCE IN THIS ENTRYTHERE IS A CLEAVED PEPTIDE FROM THE SAME PROTEIN (CHAIN A)ASSOCIATED WITH THE TRANSPEPTIDASE DOMAIN OFPBP-1A (CHAIN B), THE ENTRY IS MARKED AS DIMERIC. |
-Components
-PENICILLIN-BINDING PROTEIN ... , 2 types, 2 molecules AB
| #1: Protein/peptide | Mass: 1825.027 Da / Num. of mol.: 1 / Fragment: GLYLOSYLTRANSFERASE DOMAIN, RESIDUES 51-66 Source method: isolated from a genetically manipulated source Source: (gene. exp.) STREPTOCOCCUS PNEUMONIAE (bacteria) / Strain: R6 / Plasmid: PJAH143 / Production host: |
|---|---|
| #2: Protein | Mass: 43179.594 Da / Num. of mol.: 1 / Fragment: TRANSPEPTIDASE DOMAIN, RESIDUES 267-650 Source method: isolated from a genetically manipulated source Source: (gene. exp.) STREPTOCOCCUS PNEUMONIAE (bacteria) / Strain: R6 / Plasmid: PJAH143 / Production host: |
-Non-polymers , 5 types, 75 molecules 


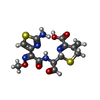

| #3: Chemical | ChemComp-ZN /  Mass: 65.409 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: Zn#4: Chemical |  Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl#5: Chemical |  Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2#6: Chemical | ChemComp-CEF / |  Mass: 397.429 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H15N5O5S2 Mass: 397.429 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H15N5O5S2#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 65 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | Y |
|---|---|
| Sequence details | EXTRA PEPTIDE FROM THE GLYLOSYLTRANSFERASE DOMAIN, RESIDUES 51-66. THE SEQUENCE IN THE SEQRES ...EXTRA PEPTIDE FROM THE GLYLOSYLTR |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 3 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.24 Å3/Da / Density % sol: 60.62 % Description: PARTIAL PHASING INFORMATION FROM DIFFERENT SOURCES WAS DEVISED USING SHARP |
|---|---|
| Crystal grow | pH: 7 / Details: 13% PEG1000,50 MM NACL, 5MM ZNSO4,50MM TRIS PH 7.0 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-4 / Wavelength: 0.9793 / Beamline: ID14-4 / Wavelength: 0.9793 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Oct 1, 2001 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9793 Å / Relative weight: 1 |
| Reflection | Resolution: 2.55→50 Å / Num. obs: 16766 / % possible obs: 88.4 % / Observed criterion σ(I): 2 / Redundancy: 3.5 % / Biso Wilson estimate: 53 Å2 / Rsym value: 0.06 / Net I/σ(I): 15.09 |
| Reflection shell | Resolution: 2.55→2.71 Å / Redundancy: 2.5 % / Mean I/σ(I) obs: 3.41 / Rsym value: 0.29 / % possible all: 54.1 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2BG4 2bg4 Resolution: 2.55→47.07 Å / Rfactor Rfree error: 0.009 / Data cutoff high absF: 4488958.94 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 39.754 Å2 / ksol: 0.342401 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 54.6 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.55→47.07 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.55→2.71 Å / Rfactor Rfree error: 0.041 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
