 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1wc3: Soluble adenylyl cyclase CyaC from S. platensis in complex with a... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1wc3 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Soluble adenylyl cyclase CyaC from S. platensis in complex with alpha, beta-methylene-ATP and Strontium | ||||||
 Components Components | ADENYLATE CYCLASE | ||||||
 Keywords Keywords | LYASE / CYCLASE / SOLUBLE ADENYLYL CYCLASE / CAMP SIGNALING | ||||||
| Function / homology |  Function and homology information Function and homology informationcyclic nucleotide biosynthetic process / adenylate cyclase activity / phosphorelay sensor kinase activity / histidine kinase / ATP binding / metal ion binding Similarity search - Function | ||||||
| Biological species |  SPIRULINA PLATENSIS (bacteria) SPIRULINA PLATENSIS (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Steegborn, C. / Litvin, T.N. / Levin, L.R. / Buck, J. / Wu, H. | ||||||
 Citation Citation | Journal: Nat.Struct.Mol.Biol. / Year: 2005 Title: Bicarbonate Activation of Adenylyl Cyclase Via Promotion of Catalytic Active Site Closure and Metal Recruitment Authors: Steegborn, C. / Litvin, T.N. / Levin, L.R. / Buck, J. / Wu, H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1wc3.cif.gz | 97.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1wc3.ent.gz | 73.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1wc3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/wc/1wc3ftp://data.pdbj.org/pub/pdb/validation_reports/wc/1wc3 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1wc0SC  1wc1C  1wc4C  1wc5C  1wc6C S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj










- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 24189.678 Da / Num. of mol.: 2 / Fragment: CATALYTIC DOMAIN, RESIDUES 1005-1202 Source method: isolated from a genetically manipulated source Source: (gene. exp.) SPIRULINA PLATENSIS (bacteria) / Production host: #2: Chemical | 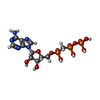  Mass: 505.208 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-CPP, energy-carrying molecule analogue*YM Mass: 505.208 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-CPP, energy-carrying molecule analogue*YM#3: Chemical |   Mass: 87.620 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Sr Mass: 87.620 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Sr#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 227 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 227 / Source method: isolated from a natural source / Formula: H2OSequence details | THE PROTEIN WAS EXPRESSED WITH N-TERMNAL HIS-TAG CONSTRUCT COMPRISING OF RESIDUES MARKED AS 984- ...THE PROTEIN WAS EXPRESSED WITH N-TERMNAL HIS-TAG CONSTRUCT COMPRISING | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.98 Å3/Da / Density % sol: 37.74 % |
|---|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X4A / Wavelength: 0.97911 / Beamline: X4A / Wavelength: 0.97911 |
| Detector | Type: ADSC CCD / Detector: CCD |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97911 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→20 Å / Num. obs: 30126 / % possible obs: 98.8 % / Observed criterion σ(I): 0 / Redundancy: 4.7 % / Biso Wilson estimate: 8.9 Å2 / Rmerge(I) obs: 0.06 / Net I/σ(I): 10.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1WC0 Resolution: 1.9→15 Å / Rfactor Rfree error: 0.006 / Cross valid method: THROUGHOUT / σ(F): 3
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 28.8 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→15 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→2.02 Å / Rfactor Rfree error: 0.018 / Total num. of bins used: 6
|
