 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1vwr: STREPTAVIDIN-CYCLO-[5-S-VALERAMIDE-HPQGPPC]K-NH2, PH 3.5, I4122 C... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1vwr | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | STREPTAVIDIN-CYCLO-[5-S-VALERAMIDE-HPQGPPC]K-NH2, PH 3.5, I4122 COMPLEX | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | COMPLEX (BIOTIN-BINDING PROTEIN/PEPTIDE) / COMPLEX (BIOTIN-BINDING PROTEIN-PEPTIDE) / CYCLIC PEPTIDE DISCOVERED BY PHAGE DISPLAY / COMPLEX (BIOTIN-BINDING PROTEIN-PEPTIDE) complex | |||||||||
| Function / homology |  Function and homology information Function and homology information | |||||||||
| Biological species |  Streptomyces avidinii (bacteria) Streptomyces avidinii (bacteria) Bothrops insularis (golden lancehead) Bothrops insularis (golden lancehead) | |||||||||
| Method |  X-RAY DIFFRACTION / Resolution: 1.5 Å X-RAY DIFFRACTION / Resolution: 1.5 Å | |||||||||
 Authors Authors | Katz, B.A. / Cass, R.T. | |||||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 1997 Title: In crystals of complexes of streptavidin with peptide ligands containing the HPQ sequence the pKa of the peptide histidine is less than 3.0. Authors: Katz, B.A. / Cass, R.T. #1: Journal: J.Am.Chem.Soc. / Year: 1996Title: Structure-Based Design Tools: Structural and Thermodynamic Comparison with Biotin of a Small Molecule that Binds Streptavidin with Micromolar Affinity Authors: Katz, B.A. / Liu, B. / Cass, R.T. #2: Journal: J.Am.Chem.Soc. / Year: 1996Title: Preparation of a Protein-Dimerizing Ligand by Topochemistry and Structure-Based Design Authors: Katz, B.A. #3: Journal: J.Biol.Chem. / Year: 1995Title: Topochemical Catalysis Achieved by Structure-Based Ligand Design Authors: Katz, B.A. / Cass, R.T. / Liu, B. / Arze, R. / Collins, N. #4: Journal: Chem.Biol. / Year: 1995Title: Topochemistry for Preparing Ligands that Dimerize Receptors Authors: Katz, B.A. / Stroud, R.M. / Collins, N. / Liu, B. / Arze, R. #5: Journal: Biochemistry / Year: 1995Title: Binding to Protein Targets of Peptidic Leads Discovered by Phage Display: Crystal Structures of Streptavidin-Bound Linear and Cyclic Peptide Ligands Containing the Hpq Sequence Authors: Katz, B.A. #6: Journal: J.Am.Chem.Soc. / Year: 1995Title: Structure-Based Design of High Affinity Streptavidin Binding Cyclic Peptide Ligands Containing Thioether Cross-Links Authors: Katz, B.A. / Johnson, C.R. / Cass, R.T. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1vwr.cif.gz | 67.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1vwr.ent.gz | 51.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1vwr.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/vw/1vwrftp://data.pdbj.org/pub/pdb/validation_reports/vw/1vwr | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1vwaC  1vwbC  1vwcC  1vwdC  1vweC  1vwfC  1vwgC  1vwhC  1vwiC  1vwjC  1vwkC  1vwlC  1vwmC  1vwnC  1vwoC  1vwpC  1vwqC C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 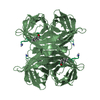
| ||||||||||||||||||
| Unit cell |
| ||||||||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 12965.025 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Streptomyces avidinii (bacteria) / References: UniProt: P22629 | ||
|---|---|---|---|
| #2: Protein/peptide | Mass: 863.017 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Bothrops insularis (golden lancehead) | ||
| #3: Chemical | ChemComp-LEA / 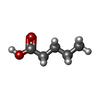  Mass: 102.132 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H10O2 Mass: 102.132 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H10O2 | ||
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 70 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 70 / Source method: isolated from a natural source / Formula: H2O | ||
| Compound details | DISULFIDE BOND OF PHAGE-DISCOVERED| Has protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.67 Å3/Da / Density % sol: 30.7 % | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 3.5 Details: SYNTHETIC MOTHER LIQUOR = 75 % SATURATED AMMONIUM SULFATE, 25 % 1.0 M POTASSIUM ACETATE ADJUSTED TO PH 3.5. | |||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / pH: 4.5 / Method: vapor diffusion, hanging drop / Details: Pahler, A., (1987) J. Biol. Chem., 262, 13933. | |||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Num. obs: 27343 / Redundancy: 4 % / Rmerge(I) obs: 0.068 |
| Reflection | *PLUS Highest resolution: 1.33 Å / Num. measured all: 108525 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.5→7.5 Å / σ(F): 2 Details: THE FOLLOWING ATOMS HAD WEAK DENSITY AND OCCUPANCIES WERE REFINED: 13, 14, 15, (MAIN CHAIN OF GLN 24), (SIDE CHAIN OF GLN 24), (N, HN, CA, HA1, HA2 OF GLY 26), (MAIN CHAIN OF ALA 35), (SIDE ...Details: THE FOLLOWING ATOMS HAD WEAK DENSITY AND OCCUPANCIES WERE REFINED: 13, 14, 15, (MAIN CHAIN OF GLN 24), (SIDE CHAIN OF GLN 24), (N, HN, CA, HA1, HA2 OF GLY 26), (MAIN CHAIN OF ALA 35), (SIDE CHAIN OF ALA 35), (CG, OD1, AND OD2 OF ASP 36), 46, 47, 48, 49, 50, (MAIN CHAIN AND CB, HB1, HB2 OF GLU 51), (CG, HG1, HG2, CD, OE1, OE2 OF GLU 51), (SIDE CHAIN OF SER 52), (TERMINUS OF ARG 53), GLY 99, ALA 100, (N AND HN OF GLU 101), (CG, HG1, HG2, CD, OE1, OE2 OF GLU 101), TERMINUS OF ARG 103, (CD, OE1, OE2 OF GLU 116), (N, HN, CA, HA OF ALA 117), (CB, HB1, HB2, HB3 OF ALA 117), LYS 121 FROM CG OUTWARD, LYS 132 SIDE CHAIN, (LYS P 9 AND NH2 P 10). RESIDUES 60 - 69 WERE REFINED IN 2 CONFORMATIONS BECAUSE UPON PROTONATION OF ASP 61 AT LOW PH, ASP 61 UNDERGOES A LARGE SHIFT IN CONFORMATION AND CHANGE IN HYDROGEN BONDING. THE LOOP COMPRISING RESIDUES 61 - 69 ALSO UNDERGO CORRESPONDING CONFORMATIONAL CHANGES. HOWEVER SOME OF THESE RESIDUES ARE DISORDERED AND NOT VISIBLE IN EITHER CONFORMATION.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.5→7.5 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.5→1.57 Å / % reflection obs: 39.7 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
