 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1v9g | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Neutron Crystallographic analysis of the Z-DNA hexamer CGCGCG | ||||||||||||||||||||
 Components Components | 5'-D(* Keywords KeywordsDNA / Z-DNA / HYDROGEN / HYDRATION / H/D EXCHANGE / NEUTRON | Function / homology | DEUTERATED WATER / N,N'-BIS(3-AMMONIOPROPYL)BUTANE-1,4-DIAMINIUM / DNA |  Function and homology information Function and homology informationBiological species | synthetic construct (others) | Method | NEUTRON DIFFRACTION / NUCLEAR REACTOR / |  MOLECULAR REPLACEMENT / Resolution: 1.8 Å MOLECULAR REPLACEMENT / Resolution: 1.8 Å  Authors AuthorsChatake, T. / Tanaka, I. / Niimura, N. |  CitationJournal: Acta Crystallogr.,Sect.D / Year: 2005 CitationJournal: Acta Crystallogr.,Sect.D / Year: 2005Title: The hydration structure of a Z-DNA hexameric duplex determined by a neutron diffraction technique. Authors: Chatake, T. / Tanaka, I. / Umino, H. / Arai, S. / Niimura, N. History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1v9g.cif.gz | 21.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1v9g.ent.gz | 14.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1v9g.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/v9/1v9gftp://data.pdbj.org/pub/pdb/validation_reports/v9/1v9g | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1woeC  1iotS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |






-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: DNA chain | Mass: 1810.205 Da / Num. of mol.: 2 / Source method: obtained synthetically / Details: Z-DNA duplex / Source: (synth.) synthetic construct (others) #2: Chemical | ChemComp-SPW / | 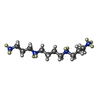  Mass: 216.434 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H20N4 Mass: 216.434 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H20N4#3: Chemical | ChemComp-DOD / | 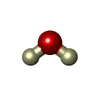  Mass: 18.015 Da / Num. of mol.: 44 / Source method: isolated from a natural source / Formula: D2O Mass: 18.015 Da / Num. of mol.: 44 / Source method: isolated from a natural source / Formula: D2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: NEUTRON DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.35 Å3/Da / Density % sol: 7.98 % | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 328 K / Method: small tubes / pH: 7 Details: MPD, sodium cacodylate, magnesium chloride, spermine, pH 7.0, SMALL TUBES, temperature 328K | ||||||||||||||||||||||||||||||||
| Components of the solutions |
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: NUCLEAR REACTOR / Site: JRR-3M  / Beamline: 1G-A / Wavelength: 2.88 Å / Beamline: 1G-A / Wavelength: 2.88 Å |
| Detector | Type: BIX-3M / Detector: DIFFRACTOMETER / Date: Jun 10, 2003 |
| Radiation | Monochromator: Elastically-bent perfect silicon monochromator Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: neutron |
| Radiation wavelength | Wavelength: 2.88 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→50 Å / Num. all: 2627 / Num. obs: 2627 / % possible obs: 73.7 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 2 % / Biso Wilson estimate: 3.9 Å2 / Rmerge(I) obs: 0.127 / Net I/σ(I): 6.2 |
| Reflection shell | Resolution: 1.6→1.66 Å / Redundancy: 1.7 % / Rmerge(I) obs: 0.208 / Mean I/σ(I) obs: 3.5 / Num. unique all: 142 / % possible all: 40.9 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1IOT Resolution: 1.8→25.05 Å / Data cutoff high absF: 3 / Data cutoff low absF: 0 / Isotropic thermal model: isotropic / Cross valid method: THROUGHOUT / σ(F): 3
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: bulk solvent correction / Bsol: 20.1 Å2 / ksol: 0.06 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 12.09 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→25.05 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: NEUTRON DIFFRACTION / Total num. of bins used: 6
|
