 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ujw: Structure of the complex between BtuB and Colicin E3 Receptor bin... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ujw | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of the complex between BtuB and Colicin E3 Receptor binding domain | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | TRANSPORT PROTEIN/HYDROLASE / beta-barrel / coiled-coil / TRANSPORT PROTEIN-HYDROLASE COMPLEX | |||||||||
| Function / homology |  Function and homology information Function and homology informationABC-type vitamin B12 transporter activity / extrachromosomal circular DNA / cobalamin transport / hydrolase activity, acting on ester bonds / porin activity / pore complex / cell outer membrane / ribosome binding / endonuclease activity / Lyases; Phosphorus-oxygen lyases ...ABC-type vitamin B12 transporter activity / extrachromosomal circular DNA / cobalamin transport / hydrolase activity, acting on ester bonds / porin activity / pore complex / cell outer membrane / ribosome binding / endonuclease activity / Lyases; Phosphorus-oxygen lyases / monoatomic ion transmembrane transport / killing of cells of another organism / transmembrane transporter binding / tRNA binding / defense response to bacterium / lyase activity / rRNA binding / protein domain specific binding / calcium ion binding / extracellular region / membrane Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.75 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.75 Å | |||||||||
 Authors Authors | Kurisu, G. / Zakharov, S.D. / Zhalnina, M.V. / Bano, S. / Eroukova, V.Y. / Rokitskaya, T.I. / Antonenko, Y.N. / Wiener, M.C. / Cramer, W.A. | |||||||||
 Citation Citation | Journal: NAT.STRUCT.BIOL. / Year: 2003 Title: The structure of BtuB with bound colicin E3 R-domain implies a translocon Authors: Kurisu, G. / Zakharov, S.D. / Zhalnina, M.V. / Bano, S. / Eroukova, V.Y. / Rokitskaya, T.I. / Antonenko, Y.N. / Wiener, M.C. / Cramer, W.A. | |||||||||
| History |
| |||||||||
| Remark 650 | HELIX DETERMINATION METHOD: AUTHOR DETERMINED |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ujw.cif.gz | 155.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ujw.ent.gz | 120.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1ujw.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/uj/1ujwftp://data.pdbj.org/pub/pdb/validation_reports/uj/1ujw | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1nqeS S: Starting model for refinement |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 2 types, 2 molecules AB
| #1: Protein | Mass: 66386.180 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|---|
| #2: Protein | Mass: 15110.648 Da / Num. of mol.: 1 / Fragment: R-domain(residues 314-448) Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
-Sugars , 1 types, 2 molecules 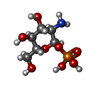
| #3: Sugar |  Type: D-saccharide / Mass: 259.151 Da / Num. of mol.: 2 Type: D-saccharide / Mass: 259.151 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Formula: C6H14NO8P |
|---|
-Non-polymers , 5 types, 28 molecules 
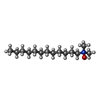
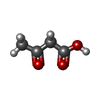


| #4: Chemical |  Mass: 256.381 Da / Num. of mol.: 2 Mass: 256.381 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Formula: C15H28O3 #5: Chemical | ChemComp-LDA /  Mass: 229.402 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C14H31NO / Comment: LDAO, detergent*YM Mass: 229.402 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C14H31NO / Comment: LDAO, detergent*YM#6: Chemical | ChemComp-AAE / |  Mass: 102.089 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O3 Mass: 102.089 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O3#7: Chemical | ChemComp-GOL /  Mass: 92.094 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C3H8O3#8: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 10 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.5 Å3/Da / Density % sol: 72.44 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS Temperature: 20 ℃ / pH: 6.5 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 19-BM / Beamline: 19-BM |
| Detector | Type: SBC / Detector: CCD |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Relative weight: 1 |
| Reflection | Resolution: 2.75→50 Å / Num. obs: 37175 |
| Reflection | *PLUS Lowest resolution: 35.5 Å / % possible obs: 96.7 % / Redundancy: 3.8 % / Num. measured all: 139699 / Rmerge(I) obs: 0.084 |
| Reflection shell | *PLUS Highest resolution: 2.75 Å / Lowest resolution: 2.85 Å / % possible obs: 91.1 % / Rmerge(I) obs: 0.295 / Mean I/σ(I) obs: 2.9 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1NQE Resolution: 2.75→37.88 Å / Cor.coef. Fo:Fc: 0.916 / Cor.coef. Fo:Fc free: 0.876 / SU B: 14.189 / SU ML: 0.275 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.469 / ESU R Free: 0.333 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 33.257 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.75→37.88 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.75→2.821 Å / Total num. of bins used: 20 /
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 35.5 Å / % reflection Rfree: 5 % | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
